I chimici delle Argonne hanno identificato un modo per convertire il cicloesano in cicloesene o cicloesadiene, entrambe importanti sostanze chimiche in un'ampia gamma di processi industriali. Il nuovo processo avviene a basse temperature, eliminando la creazione di sottoprodotti indesiderati. Credito:Laboratorio nazionale Argonne
I chimici impiegano una grande quantità di tempo ed energia cercando di far iniziare o accelerare le reazioni chimiche, ma a volte può essere altrettanto importante fermarle prima che si spingano troppo oltre.
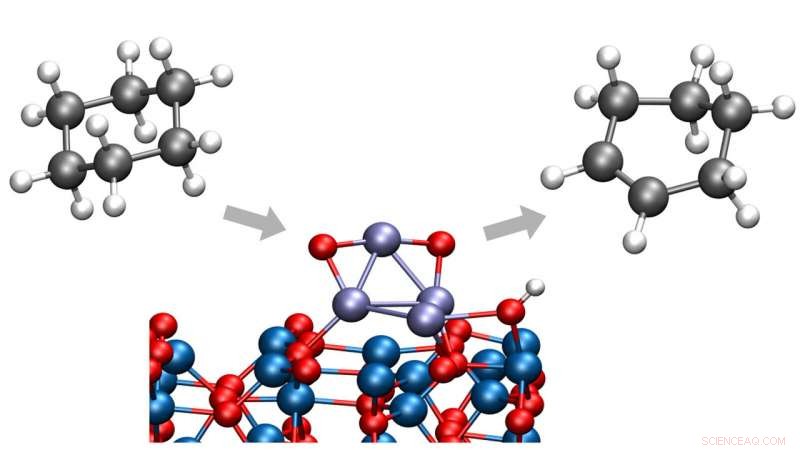
In un recente studio dell'Argonne National Laboratory del Dipartimento dell'Energia degli Stati Uniti (DOE), i chimici hanno identificato un modo per convertire il cicloesano in cicloesene o cicloesadiene, sostanze chimiche importanti in un'ampia gamma di processi industriali. È importante sottolineare che questo processo avviene a basse temperature, eliminando la creazione di anidride carbonica che sarebbe derivata da una rottura indesiderata dei legami carbonio-carbonio.
Il cicloesano è un'importante molecola di partenza in un'ampia gamma di reazioni chimiche, secondo il chimico delle Argonne Stefan Vajda, ora presso l'Istituto di chimica fisica J. Heyrovský a Praga. Però, senza un catalizzatore adatto per avviare la reazione, la conversione del cicloesano in prodotti utili richiede tipicamente temperature elevate generate attraverso il dispendio di una grande quantità di energia, e il processo può anche soffrire di scarsa selettività.
Nello studio, Il chimico Vajda e Argonne Larry Curtiss e il loro team internazionale di collaboratori hanno esaminato un tipo di reazione chiamata deidrogenazione ossidativa, in cui le molecole di idrogeno vengono strappate da una molecola più grande. Tagliando un numero limitato di legami idrogeno-carbonio, la reazione può produrre cicloesene e cicloesadiene prima che avvenga la combustione ad anidride carbonica.
Il lavoro ha migliorato gli studi precedenti del team Argonne sulla deidrogenazione di cicloesano e cicloesene introducendo due componenti chiave:un catalizzatore di ossido di cobalto di dimensioni sub-nanometriche su un supporto di ossido di alluminio e un ambiente di ossigeno controllato.
I ricercatori hanno impiegato tecniche di diffusione dei raggi X presso l'Advanced Photon Source (APS) di Argonne, una struttura per gli utenti dell'Office of Science del DOE, monitorare la natura e la stabilità dei catalizzatori durante il test catalitico dei cluster in tempo reale. Hanno scoperto che i cluster effettuavano la deidrogenazione parziale del cicloesano a temperature intorno ai 100 gradi Celsius, molto inferiori a quelle osservate in precedenza per questo tipo di reazione, e i cluster hanno mantenuto la loro natura ossidata e stabilità a temperature di reazione fino a 300°C.
"Il fatto che possiamo realizzare questa conversione a temperature più basse protegge i prodotti di deidrogenazione intermedi cicloesene e cicloesadiene dall'ulteriore conversione in prodotti indesiderati, " Ha detto Vajda.
Vajda e Curtiss hanno notato che il catalizzatore altamente selettivo è di lunga durata e non viene avvelenato o degradato dalla reazione. Nelle indagini teoriche e sperimentali sulla dimensione del catalizzatore, i ricercatori hanno scoperto che i gruppi di dimensioni quattro e ventisette atomi erano più o meno ugualmente efficienti nello svolgimento della reazione. "Sembra che finché il catalizzatore è di dimensioni inferiori a circa un nanometro, questa composizione funziona bene, un fattore importante per il potenziale aumento di scala di questa classe di catalizzatori da parte di catalizzatori più tradizionali, sebbene meno selettivo in termini di dimensioni, vie di sintesi." disse Vajda.
Per comprendere meglio i meccanismi di base dietro l'attività e la selettività dei catalizzatori di cobalto, i ricercatori hanno utilizzato calcoli della teoria del funzionale della densità per modellare i percorsi di reazione. "Le eccellenti prestazioni dei cluster di cobalto possono essere spiegate da calcoli teorici, che rivelano atomi di cobalto altamente attivi nei cluster e mostrano che la natura ossidata dei cluster provoca la formazione a bassa temperatura del prodotto, "Spiega Curtiss.