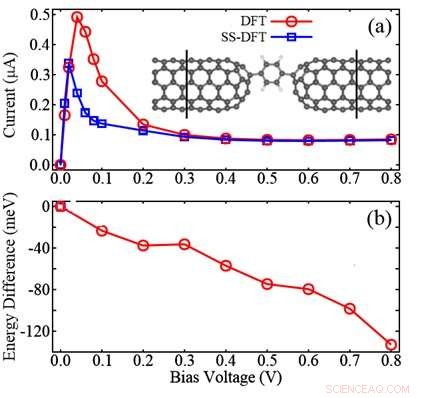
La figura mostra il confronto tra SS-DFT e metodi DFT ampiamente utilizzati per un dispositivo molecolare costituito da due elettrodi di nanotubi di carbonio (CNT) e una molecola di benzene tra:(a) curve I-V calcolate; (b) la differenza di energia calcolata sottraendo l'energia DFT da quella SS-DFT. La figura mostra che SS-DFT prevede lo stato di trasporto energeticamente più stabile con correnti elettriche inferiori rispetto al metodo basato su DFT. Credito:Zhang Chun
Gli scienziati computazionali del NUS hanno sviluppato una nuova versione della teoria del funzionale della densità (DFT) per studiare i dispositivi su scala nanometrica.
I dispositivi elettronici stanno diventando più piccoli e incorporano maggiori funzionalità. Ciò è reso possibile dalla riduzione delle dimensioni dei componenti elettronici. Quando le loro dimensioni diminuiscono, le proprietà di questi dispositivi molecolari diventano molto più sensibili al loro ambiente esterno. Sono necessari metodi computazionali per simulare e prevedere le proprietà di dispositivi così piccoli. Uno di questi è la teoria del funzionale della densità. Questi metodi sono sviluppati da principi primi, comprendente conoscenze di base e fondamentali che già conosciamo. Gli scienziati computazionali del NUS hanno perfezionato la teoria per tenere conto degli effetti di non equilibrio presenti durante il funzionamento dei dispositivi (ad esempio quando una batteria è collegata a un sistema quantistico). Questo porta a una previsione più accurata e precisa.
Il professor ZHANG Chun e il suo dottorato di ricerca. alunno, LIU Shuanglong insieme al ricercatore, Dott. Argo NURBAWONO, del Dipartimento di Fisica, NUS ha sviluppato una versione più generale della teoria del funzionale della densità (DFT) popolare e ampiamente utilizzata che può essere applicata a situazioni di non equilibrio in stato stazionario. Hanno introdotto un ulteriore grado di libertà, nota come densità elettronica di non equilibrio, nella modellazione dei primi principi. Ciò tiene conto degli effetti di non equilibrio indotti da bias quando un dispositivo molecolare opera sotto un bias finito. Questa nuova versione della teoria è nota come DFT allo stato stazionario (SS-DFT).
I ricercatori hanno dimostrato che la DFT ampiamente utilizzata in linea di principio non è corretta in uno scenario di non equilibrio in stato stazionario. In una situazione del genere, due parametri diversi, la densità elettronica totale e la densità degli elettroni che trasportano corrente, sono necessari per determinare le proprietà del corrispondente sistema di non equilibrio. La nuova teoria è stata implementata nel pacchetto computazionale SIESTA (Iniziativa spagnola per le simulazioni elettroniche con migliaia di atomi) per studiare le proprietà elettroniche/di trasporto di vari dispositivi su scala molecolare.
SS-DFT fornisce uno strumento teorico affidabile per la comprensione e la progettazione futura di nuovi dispositivi su scala molecolare con funzionalità avanzate. Il pacchetto computazionale basato su SS-DFT è ora utilizzato da molti gruppi di ricerca in tutto il mondo. Viene utilizzato per spiegare intriganti fenomeni di trasporto osservati sperimentalmente a livello molecolare e per progettare nuovi tipi di dispositivi molecolari.
I ricercatori intendono espandere l'applicabilità della teoria includendo altri effetti fisici, come le interazioni elettrone-fonone e l'accoppiamento spin-orbitale. Intendono anche migliorare l'efficienza computazionale in modo che possa essere utilizzata per modellare sistemi di grandi dimensioni intorno a 1, 000 atomi.