Capire cosa fa brillare alcune proteine richiede una comprensione della chimica. Credito:eLife - il giornale, CC BY-SA
L'intelligenza artificiale ha cambiato il modo in cui viene fatta la scienza, consentendo ai ricercatori di analizzare le enormi quantità di dati generati dai moderni strumenti scientifici. Può trovare un ago in un milione di pagliai di informazioni e, utilizzando il deep learning, può imparare dai dati stessi. L'IA sta accelerando i progressi nella ricerca genetica, nella medicina, nella progettazione di farmaci e nella creazione di composti organici.
Il deep learning utilizza algoritmi, spesso reti neurali addestrate su grandi quantità di dati, per estrarre informazioni da nuovi dati. È molto diverso dall'informatica tradizionale con le sue istruzioni dettagliate. Piuttosto, impara dai dati. Il deep learning è molto meno trasparente della tradizionale programmazione per computer, e lascia importanti domande:cosa ha imparato il sistema, cosa sa?
Come professore di chimica mi piace progettare test che abbiano almeno una domanda difficile che estenda le conoscenze degli studenti per stabilire se possono combinare idee diverse e sintetizzare nuove idee e concetti. Abbiamo ideato una domanda del genere per il figlio poster dei sostenitori dell'IA, AlphaFold, che ha risolto il problema del ripiegamento delle proteine.
Ripiegamento proteico
Le proteine sono presenti in tutti gli organismi viventi. Forniscono struttura alle cellule, catalizzano reazioni, trasportano piccole molecole, digeriscono il cibo e fanno molto di più. Sono costituiti da lunghe catene di amminoacidi come perline su un filo. Ma affinché una proteina svolga il suo lavoro nella cellula, deve torcersi e piegarsi in una complessa struttura tridimensionale, un processo chiamato ripiegamento proteico. Le proteine mal ripiegate possono portare a malattie.
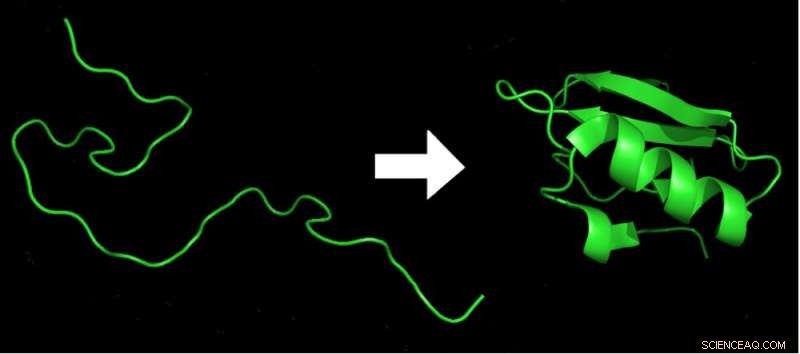
Entro pochi millisecondi dall'uscita di una catena di amminoacidi (a sinistra) dal ribosoma, viene piegata nella forma 3D a più bassa energia (a destra), necessaria per la funzione della proteina. Credito:Marc Zimmer, CC BY-ND
Nel suo discorso di accettazione del Nobel per la chimica nel 1972, Christiaan Anfinsen ha postulato che dovrebbe essere possibile calcolare la struttura tridimensionale di una proteina dalla sequenza dei suoi elementi costitutivi, gli amminoacidi.
Proprio come l'ordine e la spaziatura delle lettere in questo articolo danno senso e messaggio, così l'ordine degli amminoacidi determina l'identità e la forma della proteina, che si traduce nella sua funzione.
A causa della flessibilità intrinseca dei blocchi costitutivi dell'amminoacido, una tipica proteina può assumere circa 10 alla potenza di 300 forme diverse. Questo è un numero enorme, più del numero di atomi nell'universo. Eppure entro un millisecondo ogni proteina in un organismo si ripiegherà nella sua forma specifica, la disposizione a più bassa energia di tutti i legami chimici che compongono la proteina. Cambia solo un amminoacido tra le centinaia di amminoacidi che si trovano tipicamente in una proteina e potrebbe ripiegarsi male e non funzionare più.
AlphaFold
Per 50 anni gli informatici hanno cercato di risolvere il problema del ripiegamento delle proteine, con scarso successo. Poi, nel 2016, DeepMind, una sussidiaria AI della società madre di Google, Alphabet, ha avviato il suo programma AlphaFold. Ha utilizzato la banca dati delle proteine come set di addestramento, che contiene le strutture determinate sperimentalmente di oltre 150.000 proteine.
I neuroni che esprimono proteine fluorescenti rivelano le strutture cerebrali di due larve di moscerino della frutta. Crediti:Wen Lu e Vladimir I. Gelfand, Feinberg School of Medicine, Northwestern University
In meno di cinque anni AlphaFold ha superato il problema del ripiegamento proteico, almeno la parte più utile di esso, ovvero la determinazione della struttura proteica dalla sua sequenza di amminoacidi. AlphaFold non spiega come le proteine si pieghino così rapidamente e accuratamente. È stata una grande vittoria per l'IA, perché non solo ha accumulato un enorme prestigio scientifico, ma è stato anche un importante progresso scientifico che potrebbe influenzare la vita di tutti.
Oggi, grazie a programmi come AlphaFold2 e RoseTTAFold, ricercatori come me possono determinare la struttura tridimensionale delle proteine dalla sequenza di amminoacidi che compongono la proteina, senza alcun costo, in un'ora o due. Prima di AlphaFold2 dovevamo cristallizzare le proteine e risolvere le strutture usando la cristallografia a raggi X, un processo che richiedeva mesi e costava decine di migliaia di dollari per struttura.
Ora abbiamo anche accesso all'AlphaFold Protein Structure Database, dove Deepmind ha depositato le strutture 3D di quasi tutte le proteine presenti negli esseri umani, nei topi e in più di 20 altre specie. Ad oggi ha risolto più di un milione di strutture e prevede di aggiungere altri 100 milioni di strutture solo quest'anno. La conoscenza delle proteine è salita alle stelle. La struttura di metà di tutte le proteine conosciute sarà probabilmente documentata entro la fine del 2022, tra cui molte nuove strutture uniche associate a nuove funzioni utili.
Pensare come un chimico
AlphaFold2 non è stato progettato per prevedere come le proteine interagirebbero tra loro, tuttavia è stato in grado di modellare il modo in cui le singole proteine si combinano per formare unità complesse di grandi dimensioni composte da più proteine. Avevamo una domanda impegnativa per AlphaFold:il suo set di addestramento strutturale gli aveva insegnato un po' di chimica? Potrebbe dire se gli amminoacidi reagirebbero tra loro, un evento raro ma importante?
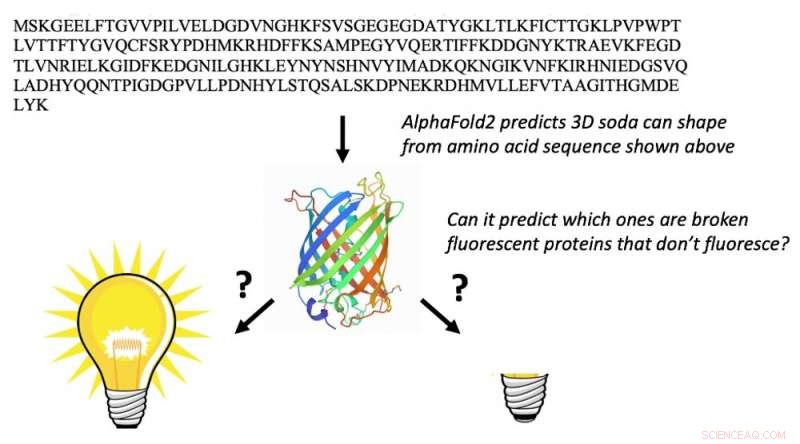
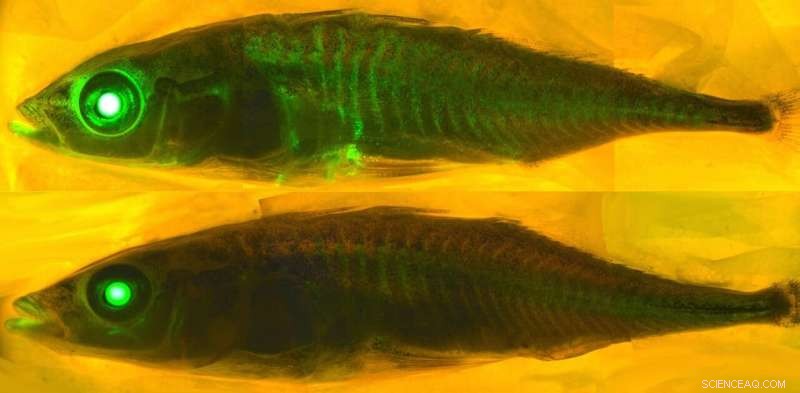
AlphaFold2 può prendere la sequenza amminoacidica delle proteine fluorescenti (lettere in alto) e prevedere le loro forme 3D a botte (al centro). Questo non è sorprendente. Ciò che è totalmente inaspettato è che può anche prevedere quali proteine fluorescenti sono "rotte" e non possono essere fluorescenti. Credito:Marc Zimmer, CC BY-ND
Sono un chimico computazionale interessato alle proteine fluorescenti. Queste sono proteine che si trovano in centinaia di organismi marini come meduse e coralli. Il loro bagliore può essere utilizzato per illuminare e studiare le malattie.
Ci sono 578 proteine fluorescenti nella banca dati delle proteine, di cui 10 sono "rotte" e non fluorescenti. Le proteine raramente attaccano se stesse, un processo chiamato modificazione post-traduzionale autocatalitica, ed è molto difficile prevedere quali proteine reagiranno con se stesse e quali no.
Solo un chimico con una notevole conoscenza delle proteine fluorescenti sarebbe in grado di utilizzare la sequenza di amminoacidi per trovare le proteine fluorescenti che hanno la giusta sequenza di amminoacidi per subire le trasformazioni chimiche necessarie per renderle fluorescenti. Quando abbiamo presentato AlphaFold2 con le sequenze di 44 proteine fluorescenti che non sono nella banca dati delle proteine, ha piegato le proteine fluorescenti fissate in modo diverso da quelle rotte.
Il risultato ci ha sbalordito:AlphaFold2 aveva imparato un po' di chimica. Aveva scoperto quali amminoacidi nelle proteine fluorescenti fanno la chimica che le fa brillare. Sospettiamo che il set di addestramento della banca dati delle proteine e gli allineamenti multipli delle sequenze consentano ad AlphaFold2 di "pensare" come i chimici e cercare gli amminoacidi necessari per reagire tra loro per rendere la proteina fluorescente.
Un programma pieghevole che impara un po' di chimica dal suo set di formazione ha anche implicazioni più ampie. Facendo le domande giuste, cos'altro si può ottenere da altri algoritmi di deep learning? Gli algoritmi di riconoscimento facciale potrebbero trovare marcatori nascosti per le malattie? Gli algoritmi progettati per prevedere i modelli di spesa tra i consumatori potrebbero anche trovare una propensione a piccoli furti o inganni? E, cosa più importante, questa capacità, e simili incrementi di abilità in altri sistemi di intelligenza artificiale, è desiderabile? + Esplora ulteriormente
Questo articolo è stato ripubblicato da The Conversation con licenza Creative Commons. Leggi l'articolo originale.