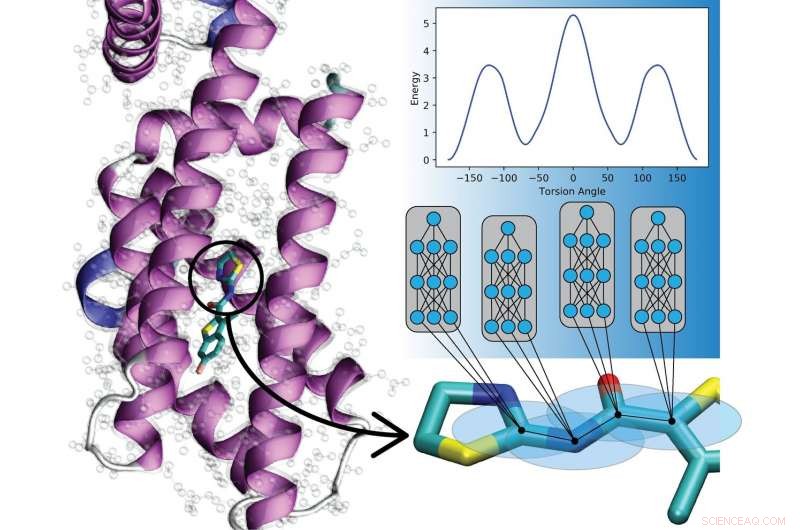
Nuovi modelli di deep learning prevedono le interazioni tra gli atomi nelle molecole organiche. Questi modelli aiuteranno i biologi computazionali e i ricercatori nello sviluppo di farmaci a comprendere e curare le malattie. Credito:Los Alamos National Laboratory
Nuovo lavoro dal Los Alamos National Laboratory, l'Università della Carolina del Nord a Chapel Hill, e l'Università della Florida sta dimostrando che le reti neurali artificiali possono essere addestrate a codificare le leggi della meccanica quantistica per descrivere i movimenti delle molecole, simulazioni di sovralimentazione potenzialmente in un'ampia gamma di campi.
"Ciò significa che ora possiamo modellare materiali e dinamiche molecolari miliardi di volte più velocemente rispetto ai metodi quantistici convenzionali, pur mantenendo lo stesso livello di precisione, " ha detto Justin Smith, Fisico di Los Alamos e Metropolis Fellow nella Divisione Teorica del laboratorio. Comprendere come si muovono le molecole è fondamentale per sfruttare il loro potenziale valore per lo sviluppo di farmaci, simulazioni proteiche e chimica reattiva, Per esempio, e sia la meccanica quantistica che i metodi sperimentali (empirici) alimentano le simulazioni.
La nuova tecnica, chiamato potenziale ANI-1ccx, promette di far progredire le capacità dei ricercatori in molti campi e migliorare l'accuratezza dei potenziali basati sull'apprendimento automatico negli studi futuri sulle leghe metalliche e sulla fisica della detonazione.
Algoritmi di meccanica quantistica (QM), utilizzato su computer classici, può descrivere accuratamente i movimenti meccanici di un composto nel suo ambiente operativo. Ma QM scala molto male con dimensioni molecolari variabili, limitando fortemente la portata delle possibili simulazioni. Anche un leggero aumento delle dimensioni molecolari all'interno di una simulazione può aumentare notevolmente il carico computazionale. Quindi i professionisti ricorrono spesso all'uso di informazioni empiriche, che descrive il moto degli atomi in termini di fisica classica e leggi di Newton, consentendo simulazioni che raggiungono miliardi di atomi o milioni di composti chimici.
Tradizionalmente, le potenzialità empiriche hanno dovuto trovare un compromesso tra accuratezza e trasferibilità. Quando i molti parametri del potenziale sono sintonizzati con precisione per un composto, la precisione diminuisce su altri composti.
Anziché, la squadra di Los Alamos, con l'Università della Carolina del Nord a Chapel Hill e l'Università della Florida, ha sviluppato un approccio di apprendimento automatico chiamato apprendimento di trasferimento che consente loro di costruire potenziali empirici imparando dai dati raccolti su milioni di altri composti. Il nuovo approccio con il potenziale empirico di apprendimento automatico può essere applicato a nuove molecole in millisecondi, consentendo la ricerca su un numero molto maggiore di composti su scale temporali molto più lunghe.