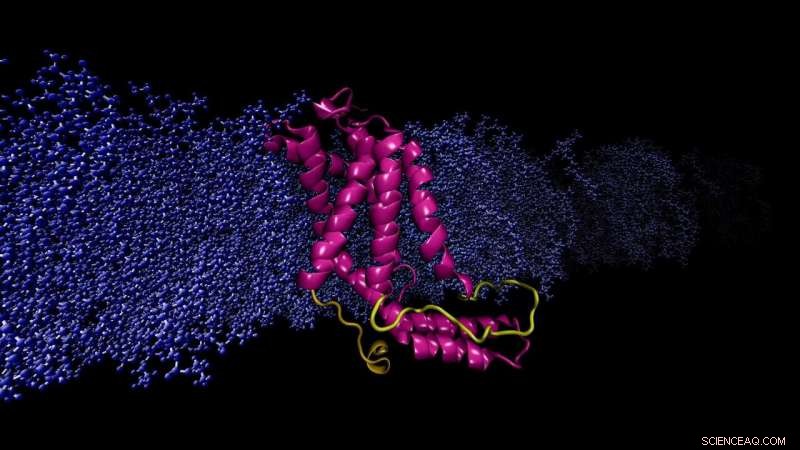
Modello computazionale Membrana cellulare incorporata nella proteina YidC2. Il ciclo modellato (giallo), mancante nella struttura cristallina a raggi X, è fondamentale per la stabilizzazione delle proteine. Credito:Sogol Moradi
Un nuovo studio condotto da chimici dell'Università dell'Arkansas mostra che la cristallografia a raggi X, il metodo standard per determinare la struttura delle proteine, può fornire informazioni imprecise su un insieme critico di proteine, quelle che si trovano nelle membrane cellulari, che a loro volta potrebbero portare a una progettazione di farmaci scadente e inefficiente.
I risultati dei ricercatori sono stati pubblicati oggi in Rapporti scientifici , una pubblicazione sulla natura.
"Due terzi di tutti i farmaci, compresi quelli usati per la chemioterapia, proteine bersaglio presenti sulle membrane cellulari, " disse Mahmoud Moradi, assistente professore di chimica e biochimica presso il J. William Fulbright College of Arts and Sciences. "Sfortunatamente, Cristallografia a raggi X, il gold standard per determinare la struttura delle proteine, ha molti limiti quando si tratta di quelli che si trovano nella membrana cellulare. Il nostro lavoro espone, e in molti modi, spiega questi limiti".
Considerate le molecole del cavallo di battaglia delle cellule, le proteine sono responsabili di quasi tutti i compiti nei sistemi viventi. Alcune proteine vivono all'interno delle cellule, e alcuni risiedono sulla membrana cellulare, uno strato esterno di lipidi che separa la cellula dal suo ambiente esterno. Le proteine di membrana sono di fondamentale importanza perché regolano lo scambio di informazioni e materiali tra la cellula e il suo ambiente, un compito vitale per la sopravvivenza e la normale funzione della cellula perché qualsiasi disturbo nella funzione proteica può provocare malattie.
Lo studio della funzione delle proteine è necessario per comprendere le basi molecolari della malattia. Per fare questo, i ricercatori si sono affidati alla cristallografia a raggi X, lo strumento principale per determinare la forma e la struttura delle proteine. La cristallografia a raggi X è essenziale anche allo scopo di progettare farmaci in grado di manipolare efficacemente la funzione delle proteine. Però, lo studio della struttura delle proteine di membrana è difficile perché il loro ambiente nativo non è compatibile con la cristallografia a raggi X. I ricercatori devono rimuovere le proteine dal loro ambiente nativo e collocarle in un ambiente lipidico artificiale prima di applicare la tecnica.
Moradi e Thomas Harkey, all'epoca studente universitario e ora studente di medicina presso l'Università dell'Arkansas per le scienze mediche, hanno affrontato questo problema da una prospettiva diversa. Da circa due anni, hanno usato un supercomputer presso l'Arkansas High Performance Computing Center per funzionare in modo continuo, calcoli a livello di microsecondi che simulano la dinamica molecolare di YidC2, una proteina di membrana con un ciclo citoplasmatico cristallograficamente irrisolto nella sua struttura molecolare. È noto che i loop citoplasmatici hanno un significato funzionale nelle proteine di membrana.
Le simulazioni di Moradi e Harkey hanno dimostrato che il ciclo citoplasmatico di YidC2 ha stabilizzato l'intera proteina, in particolare la regione C1, un'area potenzialmente importante per la progettazione di farmaci. I gruppi di testa lipidici altamente polari o carichi hanno interagito con e stabilizzato il ciclo. Questa scoperta ha dimostrato che i loop irrisolti delle proteine di membrana potrebbero essere importanti per la stabilizzazione delle proteine, nonostante l'apparente mancanza di struttura molecolare.
"Tipicamente, se parte di una proteina non viene risolta nella cristallografia a raggi X, viene interpretato come privo di una struttura particolare, "Ha detto Moradi. "Mostriamo che per le proteine di membrana e in particolare le parti della proteina che interagiscono con la membrana cellulare, questa interpretazione non è accurata e potrebbe essere fuorviante. Pensiamo che la spiegazione alternativa per il disturbo potrebbe essere che la proteina non è studiata nel suo ambiente di membrana nativo".
Moradi ha affermato che i loro risultati hanno anche dimostrato che la chimica computazionale e la tecnologia del supercalcolo possono essere utilizzate per modellare le proteine di membrana in modo più accurato in un ambiente che imita il loro ambiente fisiologico.