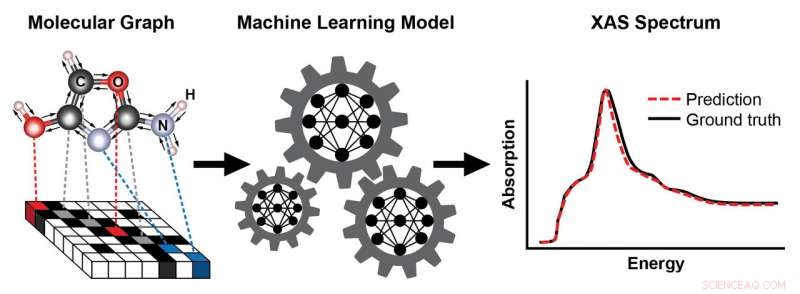
Uno schema che mostra i passaggi per addestrare un modello di apprendimento automatico per prevedere uno spettro di assorbimento dei raggi X (XAS) basato sulla struttura nota di una molecola. La struttura della molecola è rappresentata come un grafico, con atomi come nodi e legami chimici come bordi. Questa rappresentazione cattura la connettività degli atomi:qui, carbonio (C), ossigeno (O), azoto (N), e idrogeno (H) e il tipo e la lunghezza dei legami chimici che li collegano. Lo spettro XAS risultante contiene ricche informazioni sull'ambiente chimico locale degli atomi assorbenti, come la loro simmetria e il numero di atomi vicini. Credito:Brookhaven National Laboratory
La spettroscopia di assorbimento dei raggi X (XAS) è una tecnica di caratterizzazione popolare per sondare la struttura atomica locale e le proprietà elettroniche di materiali e molecole. Poiché gli atomi di ciascun elemento assorbono i raggi X ad energie caratteristiche, XAS è adatto per mappare la distribuzione spaziale degli elementi in un campione. Tipicamente, gli scienziati eseguono esperimenti XAS su sorgenti di luce di sincrotrone, come la National Synchrotron Light Source II (NSLS-II), perché forniscono una luce molto brillante, raggi X sintonizzabili. Misurando l'assorbanza in un campione a diverse energie dei raggi X, gli scienziati possono generare un grafico chiamato spettro di assorbimento dei raggi X.
"XAS è una funzionalità chiave per gli utenti dell'NSLS-II del Brookhaven National Laboratory e del Center for Functional Nanomaterials (CFN), entrambe le strutture per gli utenti dell'Ufficio delle scienze del Dipartimento dell'energia degli Stati Uniti (DOE) aperte alla comunità della ricerca scientifica, " disse Deyu Lu, un fisico nel CFN Theory and Computation Group. "Con i giusti strumenti di analisi, XAS può fornire informazioni straordinarie nella ricerca sulle nanoscienze. Lo sviluppo di tali strumenti è fondamentale per la nostra missione di strutture per gli utenti".
Classificazione degli ambienti chimici locali
Diverse regioni dello spettro di assorbimento dei raggi X sono sensibili a diversi aspetti delle proprietà del materiale in un campione. Per esempio, la struttura vicino al bordo di assorbimento dei raggi X (XANES) si concentra sulla regione del bordo vicino dello spettro, proprio sopra l'energia di insorgenza sufficiente per eccitare un elettrone dai gusci interni di un atomo a uno stato vuoto. XANES codifica informazioni dettagliate sull'ambiente chimico locale degli atomi assorbenti in un campione, inclusa la loro coordinazione geometrica, simmetria, e stato di carica (il numero di elettroni acquisiti o persi dal legame chimico). Ma analizzare i dati spettrali è molto impegnativo a causa della loro natura astratta.
"A differenza di un'immagine al microscopio di un materiale in cui è possibile vedere direttamente caratteristiche come cristallinità o difetti, Gli spettri XANES codificano informazioni che richiedono competenze di dominio per interpretare, " ha spiegato Lu.
L'interpretazione standard dei segnali in uno spettro XANES si basa su caratteristiche note come "impronte digitali, " che sono costruiti da misurazioni su materiali di riferimento. Tuttavia, questo approccio dell'impronta digitale fallisce quando il campione non è un semplice cristallo e i materiali di riferimento pertinenti non possono essere facilmente identificati.
Le simulazioni su larga scala basate sulla teoria da modelli di struttura atomica possono fornire informazioni molto utili per l'interpretazione degli spettri XANES sperimentali; però, queste simulazioni sono spesso computazionalmente costose e richiedono tempo, e il loro livello di accuratezza dipende fortemente dalle approssimazioni teoriche scelte e dal sistema in studio. Di conseguenza, interpretazione spettrale robusta è attualmente il collo di bottiglia degli studi XAS. Per di più, l'interpretazione in tempo reale degli spettri XAS è emersa come una nuova sfida per gli studi sull'evoluzione dinamica dei materiali in condizioni operative e per la sperimentazione autonoma. La necessità di robusti, l'interpretazione spettrale efficiente sta diventando sempre più diffusa alle sorgenti di luce di sincrotrone.
"Tempo reale, interpretazione accurata della diffusione dei raggi X e delle misurazioni spettroscopiche come l'assorbimento dei raggi X, fluorescenza, e la diffrazione è una capacità importante per gli utenti che conducono ricerche presso NSLS-II e altre strutture a luce di sincrotrone, " disse Mehmet Topsakal, un associato scientifico nel Materials for Energy Applications Group del dipartimento di scienza e tecnologia nucleare di Brookhaven che sta sviluppando analisi avanzate dei dati e tecniche di apprendimento automatico per la spettroscopia a raggi X. "Ogni anno, migliaia di scienziati da tutto il mondo vengono a NSLS-II per sondare le proprietà di vari materiali. Una pipeline di analisi spettrale all'avanguardia consentirebbe agli utenti di ottenere un feedback utile sui loro campioni mentre gli esperimenti sono in corso e di apportare modifiche al volo per guidare gli esperimenti. La domanda è, come possiamo fare un'interpretazione spettrale in tempo reale per scoprire le correlazioni struttura-spettro?"
Estrazione di informazioni con l'apprendimento automatico
Sfruttando i big data e l'apprendimento automatico, Lu e Topsakal hanno cercato di rispondere a questa domanda con lo scienziato computazionale Shinjae Yoo della Computational Science Initiative (CSI) del Brookhaven Lab e il dottorato di ricerca della Columbia University. candidato e DOE Computational Science Graduate Fellow Matthew Carbone.
"La borsa di studio per laureati in scienze computazionali DOE mi ha offerto un'opportunità unica di andare oltre il mio dottorato di ricerca in fisica chimica alla Columbia per esplorare la potenza degli algoritmi di apprendimento automatico, lavorando a fianco degli scienziati di Brookhaven, " ha affermato Carbone. "Il machine learning sfrutta enormi set di dati per costruire modelli altamente percettivi che, una volta addestrato, può fare previsioni al volo su nuovi dati. Tali modelli potrebbero essere utilizzati per aggirare costosi calcoli di chimica quantistica e supportare la caratterizzazione del materiale operando".
I membri di questo team e i collaboratori hanno lavorato su mappature spettro-struttura e struttura-spettro per diversi anni. Nel 2017, hanno sviluppato modelli di apprendimento automatico per prevedere i numeri medi di coordinazione delle nanoparticelle metalliche dagli spettri XANES. L'anno scorso, hanno creato un database XANES per risolvere la struttura locale di un rivestimento amorfo in ossido di titanio per applicazioni fotocatalitiche. Hanno anche costruito un modello di apprendimento automatico in grado di prevedere la simmetria locale degli atomi assorbitori da spettri XANES simulati di ossidi di metalli di transizione.
"Quando si esegue l'interpretazione spettrale basata sull'esperienza del dominio, tendiamo a concentrarci su caratteristiche specifiche progettate dalla nostra intuizione, " ha detto Lu. "Il machine learning può estrarre le informazioni di cui abbiamo bisogno in un modo statisticamente saliente che elimina i pregiudizi umani".
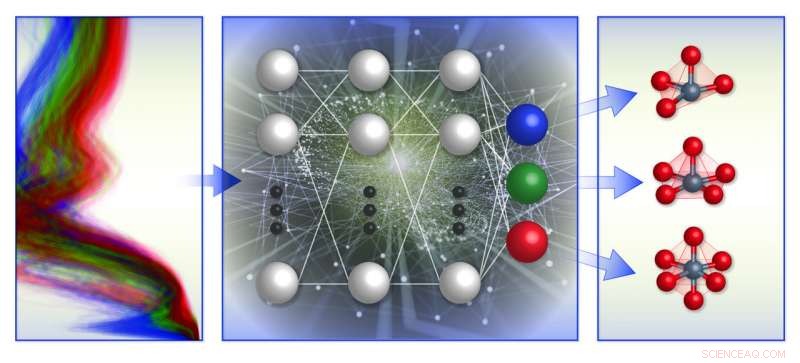
Un'illustrazione schematica del quadro di classificazione dell'ambiente chimico locale basato sullo spettro del team. Hanno addestrato i modelli di apprendimento automatico (al centro) con il database computazionale degli spettri di assorbimento dei raggi X (a sinistra) per prevedere la geometria locale attorno agli ioni di metalli di transizione carichi positivamente (a destra). Credito:Brookhaven National Laboratory
Previsione degli spettri di assorbimento dei raggi X
Basandosi sui loro successi passati, il team ha affrontato un problema più impegnativo:addestrare un modello di apprendimento automatico per prevedere rapidamente gli spettri basati su strutture molecolari note. Un tale modello eviterebbe la necessità di simulazioni computazionalmente costose, che non sono fattibili durante gli esperimenti operando, quando gli scienziati studiano i materiali in condizioni operative. Nonostante i crescenti sforzi di apprendimento automatico per prevedere le proprietà chimiche dei materiali, previsioni dirette delle funzioni spettrali dei materiali reali non erano ancora state ottenute.
"Una difficoltà tecnica è costruire una rappresentazione ottimale delle strutture molecolari in grado di codificare la simmetria intrinseca delle molecole come caratteristiche di input per il modello di apprendimento automatico, " disse Yo.
Adottando una recente idea proposta dagli scienziati di Google, Topsakal e Carbone hanno costruito un modello di apprendimento automatico basato su una rappresentazione grafica di molecole come input, dove gli atomi sono rappresentati come nodi e i legami chimici come bordi.
"I computer non possono vedere le molecole come noi, " ha detto Topsakal. "Un grafico è un modo naturale per codificare la struttura e la connettività di una molecola, catturando quali atomi sono collegati e il tipo e la lunghezza dei legami chimici che li collegano. Inoltre, questa rappresentazione è invariante a trasformazioni come traslazioni e rotazioni. Questo concetto è analogo a quello del riconoscimento delle immagini, dove un oggetto come un gatto o un cane in uno sfondo può ancora essere classificato correttamente dopo che l'immagine è stata trasformata."
Per addestrare il modello per una dimostrazione di prova di principio, il team ha utilizzato un database ben consolidato (chiamato QM9) contenente informazioni strutturali e chimiche calcolate su 134, 000 piccole molecole con fino a nove atomi pesanti per tipo di atomo (carbonio, azoto, ossigeno, e fluoro). Da questo database, hanno selezionato due sottoinsiemi di addestramento:un sottoinsieme con molecole contenenti almeno un atomo di ossigeno, e un altro sottoinsieme con molecole contenenti almeno un atomo di azoto e ha calcolato i corrispondenti spettri XANES. Quindi, hanno usato i loro modelli addestrati per prevedere gli spettri XANES per i bordi di assorbimento dell'ossigeno e dell'azoto corrispondenti alle eccitazioni degli elettroni nel guscio più interno dei rispettivi atomi.
Il modello di apprendimento automatico ha riprodotto quasi tutti i picchi di assorbimento significativi e ha previsto le posizioni dei picchi (energie a cui compaiono i picchi) e le altezze (intensità di assorbimento) con un'accuratezza molto elevata. Il modello ha anche rilevato automaticamente la conoscenza del dominio che la spettroscopia di assorbimento dei raggi X è sensibile ai gruppi funzionali, o gruppi di atomi con proprietà chimiche e reattività simili. A seconda del gruppo funzionale a cui appartiene l'atomo assorbente, caratteristiche diverse appaiono negli spettri.
"Siamo i primi a dimostrare che un modello di apprendimento automatico può essere utilizzato per prevedere con precisione le funzioni spettrali complete dei sistemi fisici reali direttamente dalle loro strutture, " ha detto Topsakal. "Anche se ci siamo concentrati sulla spettroscopia di assorbimento dei raggi X nel nostro studio, questo metodo potrebbe essere generalizzato per prevedere le informazioni spettrali per altre tecniche popolari, compresa la spettroscopia a raggi infrarossi e gamma."
"Una volta addestrato il modello di machine learning, non è necessario eseguire lunghe simulazioni fisiche, che richiedono minuti, ore, o anche giorni, " ha affermato Yoo. "Abbiamo consentito non solo la previsione degli spettri in tempo reale, ma anche la generazione simultanea di centinaia e migliaia di inferenze di spettri utilizzando più unità di elaborazione grafica, o GPU. Tale tecnologia è fondamentale per consentire controlli automatizzati della linea di luce e accelerare la scoperta scientifica. In combinazione con metodi per campionare le strutture dei materiali, tali modelli possono essere utilizzati per vagliare rapidamente le strutture rilevanti per guidare la progettazione e la scoperta dei materiali".
Prossimo, il team vorrebbe combinare i concetti del loro modello che predice la simmetria locale dagli spettri XANES e questo nuovo modello che predice gli spettri XANES dalle strutture molecolari. In definitiva, il loro obiettivo è estrarre informazioni più complete sull'ambiente chimico locale o persino sulla struttura di intere molecole da misurazioni sperimentali.
"Strumenti di apprendimento automatico, come quelli per il riconoscimento di immagini e parole e la scoperta di farmaci, sono in rapido sviluppo, " ha detto Lu. "La chiave è capire come adattare questi strumenti in modo innovativo per affrontare i problemi della scienza dei materiali".
"Il nostro obiettivo nello sviluppo di tecnologie di intelligenza artificiale e apprendimento automatico è quello di risolvere sfide scientifiche uniche sia adottando le ultime scoperte tecnologiche in queste aree sia proponendo nuovi approcci che contribuiscano alle rispettive comunità di ricerca, "aggiunse Yoo.