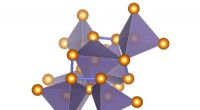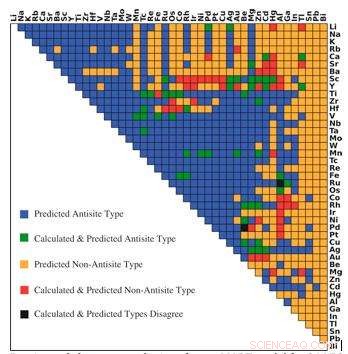
Previsione del tipo di difetto dominante dal modello r-MART per 946 intermetallici di tipo B2. I colori indicano la relazione tra previsione e calcoli come mostrato nella legenda. Credito:Bharat Medasani, Berkeley Lab / PNNL
Per la prima volta, i ricercatori del Lawrence Berkeley National Laboratory (Berkeley Lab) hanno costruito e addestrato algoritmi di apprendimento automatico per prevedere il comportamento dei difetti in alcuni composti intermetallici con elevata precisione. Questo metodo accelererà la ricerca di nuove leghe avanzate e nuovi materiali leggeri per applicazioni che vanno dall'automotive all'aerospaziale e molto altro ancora.
I loro risultati sono stati pubblicati nel numero di dicembre 2016 di Materiali computazionali naturali .
I materiali non sono mai chimicamente puri e strutturalmente impeccabili. Contengono quasi sempre dei difetti, che svolgono un ruolo importante nel dettare le proprietà. Questi difetti possono apparire come posti vacanti, che sono essenzialmente "buchi" nella struttura cristallina della sostanza, o difetti antisito, che sono essenzialmente atomi posti nel sito di cristallo sbagliato. La comprensione di tali difetti puntuali è fondamentale per gli scienziati che progettano i materiali perché possono avere un effetto drammatico sulla stabilità strutturale e sulla resistenza a lungo termine.
Tradizionalmente, i ricercatori hanno utilizzato un metodo computazionale quantomeccanico noto come calcoli funzionali della densità per prevedere quali tipi di difetti possono essere formati in una data struttura e come influenzano le proprietà del materiale. Sebbene efficace, questo approccio è molto costoso dal punto di vista computazionale da eseguire per i difetti puntuali che limitano la portata di tali indagini.
"I calcoli funzionali della densità funzionano bene se si modella una piccola unità, ma se vuoi rendere la tua cella di modellazione più grande, la potenza di calcolo necessaria per farlo aumenta sostanzialmente, "dice Bharat Medasani, un ex postdoc del Berkeley Lab e autore principale del documento npj. "E poiché è computazionalmente costoso modellare i difetti in un singolo materiale, fare questo tipo di modellazione della forza bruta per decine di migliaia di materiali non è fattibile".
Per superare queste sfide informatiche, Medasani e i suoi colleghi hanno sviluppato e addestrato algoritmi di apprendimento automatico per prevedere i difetti puntuali nei composti intermetallici, concentrandosi sulla struttura cristallina B2 ampiamente osservata. Inizialmente, hanno selezionato un campione di 100 di questi composti dal database del progetto sui materiali e hanno eseguito calcoli del funzionale della densità su supercomputer presso il National Energy Research Scientific Computing Center (NERSC), un DOE Office of Science User Facility presso il Berkeley Lab, per identificare i loro difetti.
Poiché avevano un piccolo campione di dati su cui lavorare, Medasani e il suo team hanno utilizzato un approccio forestale chiamato aumento del gradiente per sviluppare il loro metodo di apprendimento automatico con un'elevata precisione. In questo approccio sono stati successivamente costruiti ulteriori modelli di apprendimento automatico e combinati con modelli precedenti per ridurre al minimo la differenza tra le previsioni dei modelli e i risultati dei calcoli del funzionale della densità. I ricercatori hanno ripetuto il processo fino a quando non hanno raggiunto un alto livello di accuratezza nelle loro previsioni.
"Questo lavoro è essenzialmente una prova del concetto. Dimostra che possiamo eseguire calcoli funzionali della densità per poche centinaia di materiali, quindi addestrare algoritmi di apprendimento automatico per prevedere con precisione i difetti puntuali per un gruppo molto più ampio di materiali, "dice Medasani, che ora è ricercatore post-dottorato presso il Pacific Northwest National Laboratory.
"Il vantaggio di questo lavoro è che ora abbiamo un approccio di apprendimento automatico computazionalmente poco costoso in grado di prevedere in modo rapido e accurato i difetti puntuali nei nuovi materiali intermetallici", afferma Andrew Canning, un Berkeley Lab Computational Scientist e coautore dell'articolo npj. "Non dobbiamo più eseguire calcoli del primo principio molto costosi per identificare le proprietà dei difetti per ogni nuovo composto metallico".
"Questo strumento ci consente di prevedere i difetti metallici in modo più rapido e affidabile, che a sua volta accelererà la progettazione dei materiali, "dice Kristin Persson, uno scienziato del laboratorio di Berkeley e direttore del progetto sui materiali, un'iniziativa volta a ridurre drasticamente il tempo necessario per inventare nuovi materiali fornendo un accesso web-based aperto a informazioni calcolate su materiali noti e previsti. Come estensione di questo lavoro è stato sviluppato un toolkit Python open source per la modellazione di difetti puntuali in semiconduttori e isolanti (PyCDT).