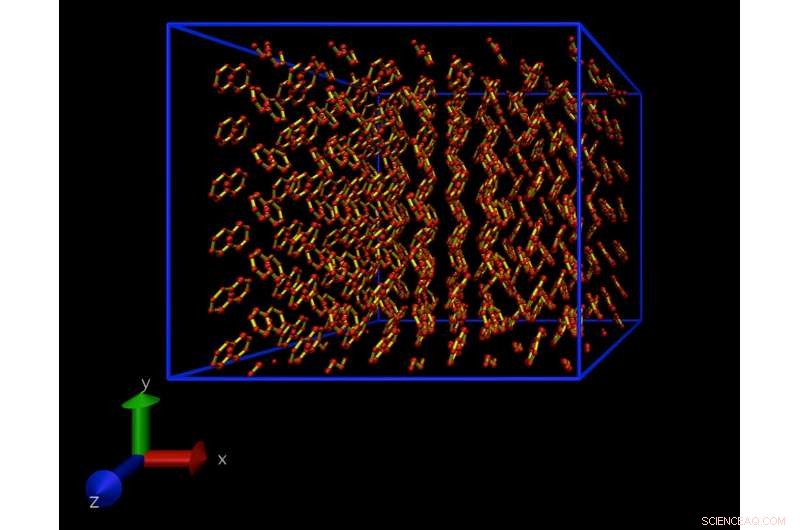
Un'istantanea da una simulazione di dinamica molecolare di un modello atomistico di un cristallo di naftalene. Questo cristallo si ripete periodicamente in tutte le direzioni, per eliminare gli effetti di superficie. Credito:Daan Frenkel, Università di Cambridge
La solubilità di una determinata sostanza, la misura di quanto bene la sostanza si dissolve in un'altra sostanza denominata solvente, dipende da proprietà di base come temperatura e pressione, così come le identità chimiche della sostanza disciolta (il soluto) e del solvente.
La previsione della solubilità è importante per una varietà di applicazioni. In campo farmaceutico, Per esempio, è fondamentale conoscere la solubilità di un farmaco poiché determina direttamente la sua disponibilità per l'organismo. L'industria petrolifera fornisce un altro esempio:le sostanze a bassa solubilità possono formare incrostazioni o depositi indesiderati nei tubi o sui trapani, causando blocchi e altri grossi problemi.
Nonostante l'importanza di prevedere la solubilità, non è cosa facile. Un approccio, utilizzando simulazioni di "forza bruta", richiede lunghi tempi di calcolo. Altre tecniche, mentre più veloce, non riescono a prevedere valori di solubilità precisi. Questa settimana in Il Giornale di Fisica Chimica , i ricercatori segnalano un nuovo tipo di software che consente stime convenienti della solubilità di praticamente qualsiasi sostanza molecolare su ampi intervalli di temperatura e pressione. Il codice fa uso di software open source prontamente disponibile e dovrebbe essere ampiamente adottato.
Daan Frenkel dell'Università di Cambridge nel Regno Unito ha lavorato con i colleghi Lunna Li, anche a Cambridge, e Tim Totton, della British Petroleum, per sviluppare il codice.
"Abbiamo fatto la scelta consapevole di utilizzare documenti ben documentati, software liberamente disponibile perché volevamo rendere disponibile il nostro approccio a chiunque, " Ha detto Frenkel. "Da molto tempo manca uno strumento di uso generale per calcolare le solubilità. La metodologia di base era lì, ma nessuno aveva effettivamente creato un programma funzionante."
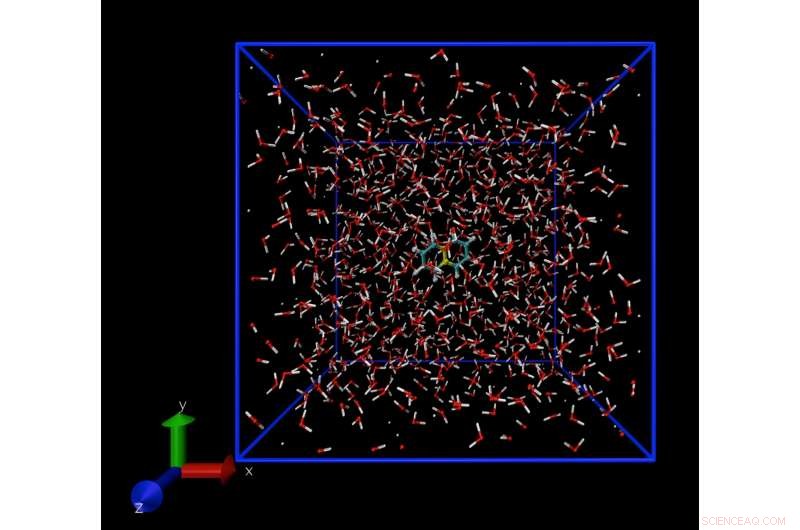
Un'istantanea di una simulazione di dinamica molecolare che mostra una singola molecola di naftalene, sciolto in acqua. La tecnica di simulazione consente di calcolare la concentrazione di molecole di naftalene in acqua al limite di solubilità. Credito:Daan Frenkel, Università di Cambridge
Il software sviluppato da questo gruppo utilizza espressioni termodinamiche standard note dalla metà del XIX secolo, come la pressione del vapore. L'approccio sfrutta il fatto che quando una fase solida o liquida sono in equilibrio, le loro pressioni di vapore sono uguali. Quando un liquido o un solido vengono riscaldati, le molecole sfuggono e formano vapore. Questa pressione di vapore può essere calcolata utilizzando modelli informatici.
Per esempio, una zolletta di zucchero che si dissolve nell'acqua:le molecole di zucchero esistono allo stato solido - la zolletta di zucchero cristallina - o completamente circondate da molecole d'acqua una volta che si sono dissolte. La quantità di zucchero in ciascuna delle due fasi, solido e soluzione, è determinata dall'energia necessaria per spostare le molecole di zucchero tra quelle fasi. La solubilità può essere calcolata calcolando la tensione di vapore delle due fasi ed equiparandole.
Per modellare la fase solida, gli investigatori hanno utilizzato un modello denominato cristallo di Einstein. In questo modello, le molecole di soluto non interagenti sono poste su un reticolo e legate a un punto reticolare con una molla matematica. La tensione di vapore del cristallo viene calcolata calcolando il lavoro necessario per spegnere le molle e attivare le interazioni tra le molecole legate.
Per modellare una molecola di soluto disciolto, i ricercatori hanno utilizzato un potenziale energetico standard per il solvente in questione, che era l'acqua negli esempi usati per testare il loro software, e ho calcolato il lavoro in tre fasi. Primo, si crea una cavità nel solvente. Una molecola di soluto viene quindi inserita nella cavità e, finalmente, la cavità è ridotta alla dimensione della molecola di soluto. Questa procedura elimina una serie di errori e produce stime accurate della tensione di vapore e, così, la solubilità.
Nel report di questa settimana, i ricercatori hanno testato il loro codice sul naftalene disciolto in acqua e hanno previsto una solubilità che si confronta bene con i valori sperimentali. Le indagini future si concentreranno sull'estensione del software in modo che possa gestire molecole di soluto più grandi.