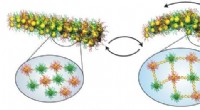
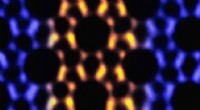
Alcuni tipi di molecole formano modelli quando vengono depositati su substrati. I dispositivi fotovoltaici e sensoriali di composti organici dipendono da questo fenomeno di auto-organizzazione. Fisici della Ludwig-Maximilians-Universitaet di Monaco di Baviera, Germania, hanno ora sviluppato un modello che prevede questi modelli e quindi consente l'ottimizzazione della sintesi molecolare in futuro.
Alcune classi di molecole sono in grado di disporsi secondo schemi specifici sulle superfici. Questa capacità di auto-organizzazione è cruciale per molte applicazioni tecnologiche, che dipendono dall'assemblaggio di strutture ordinate su superfici. Però, finora è stato virtualmente impossibile prevedere o controllare il risultato di tali processi.
Ora un gruppo di ricercatori guidati dalla dott.ssa Bianca Hermann, un fisico del Center for Nanoscience (CeNS) della LMU di Monaco, riporta una svolta significativa:combinando fisica statistica e simulazioni dettagliate con immagini ottenute mediante microscopia a effetto tunnel (STM), il team è stato in grado di formulare un modello semplice in grado di prevedere i modelli osservati. "Con l'aiuto del modello, possiamo generare un'ampia varietà di modelli che riproducono sorprendentemente bene gli arrangiamenti osservati sperimentalmente", dice Hermann. "Vogliamo estendere questo approccio ad altre simmetrie superficiali. Già ora le aree dell'elettronica molecolare, applicazioni di sensori, la catalisi superficiale e il fotovoltaico organico possono trarre vantaggio dal nostro modello. La sua capacità di prevedere le strutture formate dall'auto-organizzazione consente l'ottimizzazione dei mattoni molecolari prima della sintesi." ( Nano lettere in linea, 16 febbraio 2010)
Quando "madre natura" fa l'ingegneria, le molecole possono auto-organizzarsi in strutture complesse - un primo passo nella formazione delle membrane, cellule e altri sistemi molecolari. Il principio di autorganizzazione, che consente un uso molto parsimonioso delle risorse, viene sfruttato anche nella produzione di superfici funzionalizzate richieste nell'elettronica molecolare, applicazioni di sensori, catalisi e componenti fotovoltaici. L'idea del processo di fabbricazione è che i componenti molecolari vengano messi in contatto con un materiale di substrato, e poi "magicamente" trovano le loro posizioni preferite nella rete molecolare desiderata. I componenti di partenza sono selezionati per mostrare caratteristiche strutturali e chimiche specifiche destinate all'applicazione prevista. Però, l'ottimizzazione degli adlayer molecolari dipende in gran parte da un approccio per tentativi ed errori, ed è quindi complicato e dispendioso in termini di tempo.
Per sviluppare il nuovo modello del sito di interazione molecolare, Il gruppo del Dr. Herrmann ha collaborato con il Priv. Doz. Il Dr. Thomas Franosch e il Professor Erwin Frey all'interno del Cluster of Excellence "Nanosystems Initiative Munich" (NIM). Il problema è stato affrontato utilizzando un approccio della fisica statistica noto come metodo Monte Carlo, che permette di condurre una simulazione al computer dettagliata sulla statistica delle interazioni molecolari. I motivi strutturali così generati sono stati confrontati con immagini sperimentali ad alta risoluzione di modelli molecolari ottenuti da STM. Marta Balbas Gembra, uno studente di dottorato, ha iniziato ogni simulazione con una rappresentazione matematica di una raccolta di centinaia di particelle orientate casualmente di conformazione definita. Queste molecole schematiche sono state poi perturbate - computazionalmente - aggiungendo energia, inducendo la popolazione ad adottare una nuova configurazione.
Utilizzando questa strategia di simulazione, si può generare una maggiore varietà di modelli di quelli che si trovano naturalmente, e molti di questi corrispondevano strettamente ai modelli molecolari reali rivelati da STM. "In un caso abbiamo effettivamente previsto un modello che è stato verificato solo successivamente con STM", riferisce il dottorando Carsten Rohr. Secondo le leggi della termodinamica, i sistemi fisici tendono ad adottare lo stato con l'energia più favorevole (cioè più bassa). I test sperimentali hanno mostrato che diverse configurazioni molecolari si interconnettono fino a quando non predomina una disposizione che ricorda le tracce dei pneumatici. E senza dubbio, l'approccio Monte Carlo aveva previsto che questa disposizione corrisponde allo stato con l'energia più bassa.
"Alla fine, siamo stati in grado di dimostrare che la geometria molecolare e alcune caratteristiche salienti codificano i motivi strutturali osservati", spiega il teorico Franosch. "Abbiamo in programma di estendere l'approccio ad altri tipi di simmetrie superficiali, ma il modello fornisce già un importante strumento teorico, perché ci aiuta a prevedere il tipo di pattern di superficie che formerà una data molecola funzionale. Ciò significa che la progettazione delle molecole può essere ottimizzata durante la fase sintetica, in modo da ottenere superfici con le caratteristiche desiderate", dice Hermann. I fisici del gruppo, che provengono da diversi background scientifici e hanno potuto mettere in comune le loro competenze per questo progetto, prevedono molteplici potenziali applicazioni per il loro modello nell'elettronica molecolare, tecnologia dei sensori, catalisi e fotovoltaico. Ulteriori possibilità includono il suo utilizzo per prevedere i risultati di altri tipi di interazioni molecolari anche su substrati parzialmente modellati.