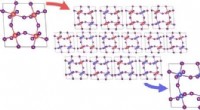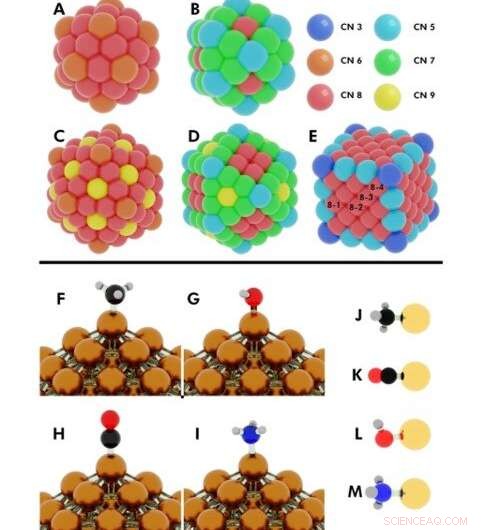
Illustrazione delle configurazioni iniziali per diversi calcoli DFT (teoria del funzionale della densità) eseguiti. Superiore:Numeri di coordinazione su (A) icosaedro a 55 atomi, (B) cubottaedro di 55 atomi, (C) icosaedro da 147 atomi, (D) cubottaedro di 147 atomi, (E) Cubo da 172 atomi. Nanoparticelle (NP) in cui più di un atomo unico condividono lo stesso numero di coordinazione (CN), sono indicati con i numeri 8-1, 8-2, 8-3, 8-4. Credito:progressi scientifici, doi:10.1126/sciadv.aax5101
Le nanoparticelle metalliche hanno ricevuto una notevole attenzione a causa delle loro applicazioni in diversi campi dalla medicina, catalisi, energia e ambiente. Però, le proprietà fondamentali dell'adsorbimento di nanoparticelle su una superficie restano da capire. James Dean e un gruppo di ricerca interdisciplinare nel dipartimento di Ingegneria Chimica, negli Stati Uniti ha introdotto un modello di adsorbimento universale per tenere conto delle caratteristiche strutturali, composizione dei metalli e diversi adsorbati di nanoparticelle tramite machine learning (ML). Il modello si adatta a un gran numero di dati per prevedere con precisione le tendenze di adsorbimento su nanoparticelle monometalliche e a base di leghe. Il modello era semplice e forniva dati calcolati rapidamente per metalli e adsorbati. Il team di ricerca ha collegato l'adsorbimento con il comportamento di stabilità per far progredire la progettazione di nanoparticelle ottimali per le applicazioni di interesse. La ricerca è ora pubblicata su Progressi scientifici .
Le nanoparticelle metalliche (NP) hanno applicazioni significative nella catalisi, che vanno dalla produzione di combustibili e prodotti chimici, all'energia solare e chimica. Ma la loro stabilità e attività catalitica mostrano generalmente tendenze opposte, dove catalizzatori molto attivi possono operare solo per pochi cicli. Una caratteristica chiave sull'estensione della funzionalità catalitica metallica dipende dalla forza di adsorbimento per una varietà di specie sulla superficie del catalizzatore. Secondo il principio Sabatier, sviluppato più di un secolo fa, i catalizzatori attivi dovrebbero legare gli adsorbati con una forza di legame che non è né forte né debole. Mentre le specie fortemente adsorbite possono avvelenare la superficie del catalizzatore, i reagenti debolmente legati si desorbono facilmente. In uno scenario intermedio, i reagenti possono incontrarsi e reagire sulle superfici catalitiche. I ricercatori attualmente utilizzano metodi di simulazione computazionale e di chimica teorica per stimolare il comportamento catalitico sui catalizzatori metallici con grande precisione per guidare i successivi esperimenti in laboratorio.
Gli sforzi computazionali si sono concentrati sullo screening di diversi catalizzatori metallici per scoprire l'energia di legame "magica" (BE) delle specie chimiche sulle superfici dei catalizzatori per formare catalizzatori molto attivi. La progettazione in silico di materiali cataliticamente attivi, però, resta da realizzare. Gli inconvenienti sono principalmente dovuti a sforzi progettuali che spesso trascurano la stabilità dei catalizzatori. I catalizzatori NP possiedono anche un alto grado di eterogeneità del sito sulla loro superficie per l'adsorbimento e la catalisi. Gli scienziati hanno sviluppato modelli di adsorbimento per mettere in relazione l'energia di legame degli adsorbati con le caratteristiche di superficie delle NP come i numeri di coordinazione (CN) per comprendere la risposta di adsorbimento specifica del sito. Ancora, per chiarezza, la variazione dell'energia di legame (BE) comporta anche descrittori secondari come la curvatura e le proprietà elettroniche delle NP.
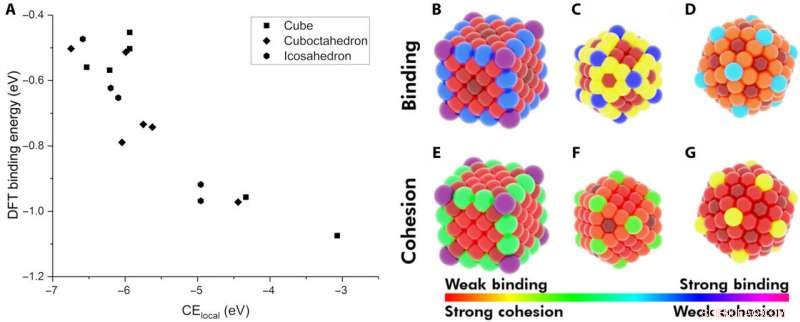
Dimostrazione dell'energia coesiva locale (CElocal) come descrittore dell'energia di adsorbimento. (A) Il BE di CO su vari siti di Au NP in funzione di CElocal:cubo di 172 atomi (rettangoli), icosaedro di 147 atomi (esagoni), e cubottaedro di 147 atomi (rombo). Mappa termica dei diversi siti sulle NP rispetto al loro BE di CO (da B a D) e al loro CElocal (da E a G). Lo schema dei colori segue l'intervallo di legame di CO più forte con CElocal più debole (viola) e di legame più debole con CElocal più forte (rosso). Credito:progressi scientifici, doi:10.1126/sciadv.aax5101
Nel presente lavoro, Dean et al. teoria del funzionale della densità applicata (DFT) e tecniche di apprendimento automatico per derivare un semplice modello basato sulla fisica per catturare con precisione l'energia di adsorbimento variabile. Hanno stimato la variabile in funzione dell'ambiente del sito di adsorbimento locale sulla superficie NP e del tipo di metallo NP. Il modello generalizzato potrebbe essere applicato a qualsiasi nanostruttura metallica per comprendere il comportamento di adsorbimento sul catalizzatore NP e la stabilità del catalizzatore; per schermare e progettare catalizzatori per numerose applicazioni.
I ricercatori hanno prima ipotizzato i fattori più importanti tra le NP monometalliche e gli adsorbati. Hanno quindi definito l'energia coesiva locale (CE Locale ) in metalli sfusi e CE catturati in NP utilizzando un modello incentrato sui legami, che ha sommato ogni energia di legame metallo-metallo. Applicando concetti simili, they described the stability of binding sites and showed how chemically unsaturated sites (fewer metal-metal bonds) bound adsorbates with an increased strength. The research team focused on describing the binding capacity of a single adsorbate-metal pair. They plotted the DFT-calculated binding energy of carbon monoxide (CO) to a 172-atom gold (Au) cube and a 147-atom gold (Au) cuboctahedron or icosahedron. The team observed a strongly inverse relationship between the local cohesive energy (CE Locale ) and binding energy (BE) to suggest the strongest adsorption sites to be those exhibiting the weakest local cohesion.
The team further developed their model and performed ordinary least squares (OLS) regression to understand adsorption on monometallic NPs and slabs using three adsorbates [Methyl radical (CH 3 ), CO, hydroxyl radical (OH)] on three different metals (Cu, Ag—silver, Au). The metallic NPs contained different morphologies (172-atom cube, 55- and 147-atom icosahedron and 55- and 147-atom cuboctahedron). They observed that the binding affinity to the adsorbates decreased as the cohesion of the local sites increased. And as the adsorbate's chemical potential increased, they became less stable and bound a metal NP with higher tendency. The direct correlation with the metal Adsorbate (MAD) intuitively described the tendency of the metal to bind the adsorbate.

Parity plot of the model-predicted binding energy (BE) of adsorbates (OH, CO, and CH3) on various metal systems versus the DFT BE (eV). (A) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which includes the nanoparticle cohesive energy (CENP) term. (B) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which does not include the CENP term. (C) The model trained on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms) and tested against RPBE (revised Perdew-Burke-Ernzerhof model) DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu). (D) The model both trained and tested on RPBE DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu) from the slab dataset. Credito:progressi scientifici, doi:10.1126/sciadv.aax5101
Dean et al. tested the generalizability of the model and trained the simulation on a single metal or single morphology, although it accurately captured other metals or morphologies as well. The work provided strong evidence that the model captured the underlying physics of the binding interactions, allowing the team to extend the work from non-periodic NPs to periodic slab systems. Computationally inexpensive systems could parameterize the model to extend to larger systems, which was not thus far possible due to the computational costs involved.
Dean et al. then extended the model from monometallic NPs to bimetallic systems. For these experiments, they plotted the BE—trained on monometallic NPs, across several sites of bi-metallic, 55-atom icosahedron NPs (Cu 31 Ag 24 and Cu 22 Ag 33 ). The model very accurately captured trends in adsorption on the bimetallic Cu/Ag NPs as well. This was an interesting result since the scientists had only trained the model on monometallic systems. The results showed the generalizability of the model for both monometallic and bimetallic NPs. Però, the team will account additional descriptors including binding site electronegativity to understand the adsorption behavior for bimetallic systems in depth.
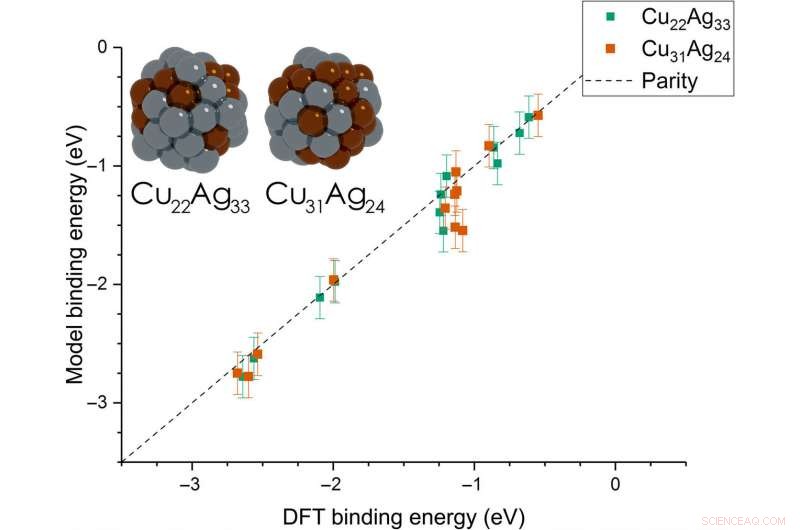
Parity plot between the presently developed model and DFT calculations on icosahedral bimetallic (Cu55−xAgx, x =24, 33) NPs. The model is trained on CH3, CO, and OH adsorbing on monometallic Ag, Cu, and Au NPs and is able to capture adsorption on bimetallic NPs. Images of the two NPs are shown as inset, with copper and silver atoms colored in brown and gray, rispettivamente. Credito:progressi scientifici, doi:10.1126/sciadv.aax5101
Although Dean et al. trained the ML (machine learning) algorithm to capture the adsorption trends of just one type of d9 metal, it could accurately predict the behavior of similar d9 metals (Cu—coper, Ag and Au). When they trained the model on a dataset of CH 3, CO and OH adsorbed to Cu, Ag and Au NPs, they could also capture general adsorption trends for similar elements in other columns of the periodic table. They then improved the complexity of the machine learning techniques to provide additional avenues to improve the model of adsorption.
In questo modo, James Dean and his colleagues developed a simple yet powerful physics-based model to capture trends on the strength of binding interactions between different adsorbates and metal NPs using machine learning techniques. The study was the first to develop an adsorption model that accurately connected the properties of diverse metal NPs with the stability of the adsorption site. The model introduced simple descriptors to capture the adsorption on any site, relative to monometallic and bimetallic NPs. The team generalized the model to effectively stimulate a range of binding interactions, including variations on the types of metals, their composition, sites of adsorption and adsorbates.
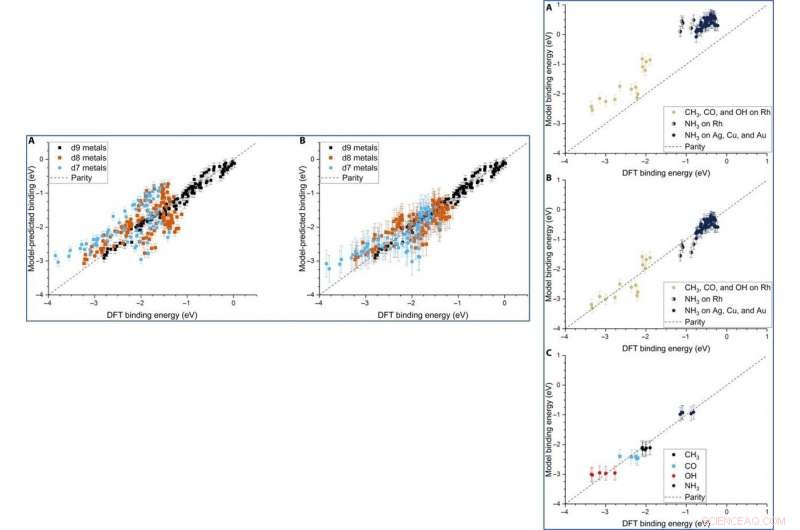
LEFT:The three-descriptor model extended to slab dataset. (A) The model trained on the slab dataset on Cu, Ag, and Au surfaces and tested against the Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au surfaces from the slab dataset. (B) The equivalent model when trained separately for each column of the d-block, still using the slab dataset. Error bars in every case are the 10-fold cross-validated RMSE of the training set. RIGHT:Extension of the model to Rh and NH3. (A) The model parameterized on our Ag, Cu, and Au NPs adsorbing CH3, CO, and OH and tested against Rh and NH3. (B) The equivalent model with empirical (constant) corrections for Rh and NH3. In the case of NH3 bound to Rh, both corrections are simultaneously applied and indicated by two-colored dots. (C) The model trained on CH3, CO, OH, and NH3 adsorbing on icosahedral/cuboctahedral Rh55. Credito:progressi scientifici, doi:10.1126/sciadv.aax5101
Although the team did not test the applicability of the model for ternary systems, the physical properties may remain relevant to accurately model multimetallic systems as well. The adsorption model can accurately describe the binding strength of a variety of molecules on any site of NPs, including alloys. The scientists expect the model to be highly applicable as a screening tool for the high throughput search of potential catalysts.
© 2019 Scienza X Rete