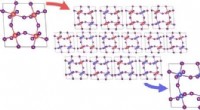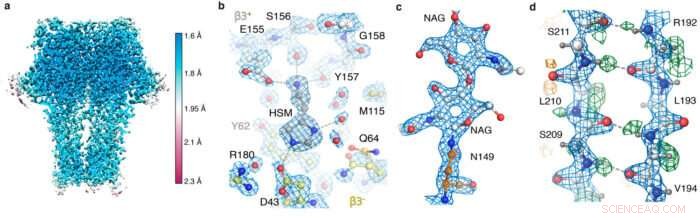
GABA UN istantanee della mappa del recettore. (a) risoluzione locale; (b) la tasca dell'agonista che mostra la coordinazione dell'istamina e le molecole d'acqua; (c) glicano legato all'N; (d) rete di legami idrogeno rivelata dalla mappa delle differenze (picchi verdi).
Osservare la precisa disposizione tridimensionale degli atomi all'interno di una proteina ci aiuta a capire come può svolgere le sue funzioni. Sebbene la criomicroscopia elettronica (crio-EM) si sia sviluppata rapidamente come importante tecnica di biologia strutturale negli ultimi anni, La cristallografia a raggi X era stata l'unica tecnica in grado di visualizzare singoli atomi. I gruppi di Radu Aricescu e Sjors Scheres al Laboratorio MRC di Biologia Molecolare, in collaborazione con scienziati di Thermo Fisher Scientific e altrove, ora sono stati in grado di risolvere singoli atomi di proteine per la prima volta in un'immagine crio-EM tridimensionale.
Questa collaborazione è iniziata all'inizio del 2019 quando Radu e Abhay Kotecha, un ricercatore presso Thermo Fisher Scientific, voleva testare il nuovo hardware crio-EM su un piccolo campione di proteine di membrana. Recettori GABAA, fulcro della ricerca di Radu da oltre un decennio, sono stati scelti perché la massima risoluzione ottenibile utilizzando la migliore tecnologia disponibile sembrava aver raggiunto un limite di circa 2,5 Ångströms (Å), ma era chiaramente necessaria una risoluzione più elevata per una migliore progettazione del farmaco.
Cos'è la risoluzione atomica?
La risoluzione è solitamente riportata in Ångströms, un'unità di lunghezza che è un decimiliardesimo di metro o 0,1 nanometri, e si riferisce alla distanza minima tra la quale due oggetti possono essere visti come separati.
La lunghezza di un tipico legame carbonio-carbonio è 1,5; altri legami nelle proteine sono un po' più corti. Così, quando la risoluzione scende a 1.2 Å, diventa possibile vedere singoli atomi all'interno di una proteina, raggiungere la vera risoluzione atomica.
Durante il test di nuovi sviluppi hardware che includevano una sorgente di elettroni con cannone a emissione di campo freddo, un nuovo filtro energetico, e una nuova fotocamera, il team ha dovuto anche sviluppare nuove strategie di elaborazione. Algoritmi per la correzione delle aberrazioni ottiche precedentemente sviluppati da Jasenko Zivanov nel gruppo di Sjors, così come un algoritmo proposto da Chris Russo e Richard Henderson, ha giocato un ruolo cruciale nello spremere la maggior parte delle informazioni dalle immagini.
Dopo aver ricevuto le immagini raccolte sul nuovo hardware del microscopio da Abhay Kotecha presso la Thermo Fisher Scientific di Eindhoven, Olanda, Takanori Nakane, un postdoc nel gruppo di Sjors, sviluppato un flusso di lavoro ottimale in RELION e Andrija Sente, insieme ad altri membri del gruppo di Radu, utilizzato questo flusso di lavoro per elaborare le immagini del recettore GABAA, durante il feedback dei risultati per ottimizzare rapidamente le impostazioni del microscopio. Una nuova, il sistema di archiviazione dati ad alta capacità sviluppato da Jake Grimmett e Toby Darling nel team di calcolo scientifico di LMB ha offerto un supporto cruciale per gestire i circa cento terabyte di dati generati. Questo sforzo di squadra sostenuto ha portato a una struttura del recettore GABAA con risoluzione di 1,7 Å senza precedenti.
Questa è stata la migliore risoluzione riportata ottenuta utilizzando cryo-EM per qualsiasi campione proteico diverso dall'apoferritina proteica. L'apoferritina è comunemente usata come punto di riferimento per crio-EM, perché la sua stabilità molecolare e la sua simmetria di 24 volte consentono ricostruzioni ad alta risoluzione da relativamente poche particelle.
Utilizzando il nuovo hardware e le strategie di elaborazione, il team è stato in grado di ottenere una struttura di apoferritina con risoluzione di 1,22 , battendo il precedente record di 1,53 per essere la struttura crio-EM a singola particella con la più alta risoluzione mai ottenuta. Più impressionante, questa risoluzione ha consentito la visualizzazione dei singoli atomi di idrogeno, anche sulle molecole d'acqua all'interno della struttura proteica. La visualizzazione delle reti di legami idrogeno all'interno delle strutture proteiche e nelle tasche di legame dei farmaci consente ai ricercatori di comprendere meglio come funzionano.
Questo lavoro rappresenta la rottura di una barriera chiave per la crio-EM come tecnica di biologia strutturale e la nuova tecnologia, raccolta dati, e le strategie di elaborazione amplieranno il numero di proteine le cui strutture possono essere risolte ad alta risoluzione. Queste ricostruzioni ad alta risoluzione consentiranno una migliore comprensione di come funzionano le proteine e faciliteranno la progettazione di farmaci più specifici che potrebbero avere un impatto sui trattamenti per una vasta gamma di malattie.