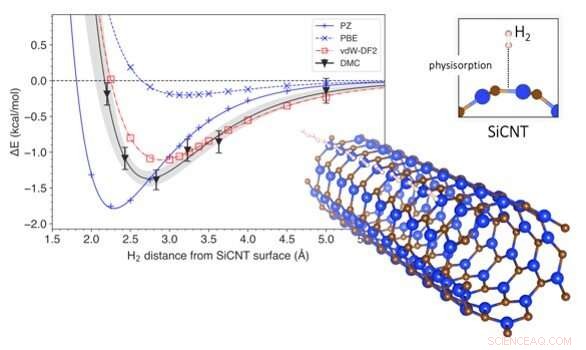
Il cambiamento di energia associato alla rimozione dell'idrogeno dai nanotubi di carburo di silicio. Il grafico mostra la variazione dell'energia del sistema con la distanza di una molecola di idrogeno dalla superficie di un nanotubo di carburo di silicio (in basso a destra). La profondità della curva indica l'energia necessaria per estrarre l'idrogeno dallo stoccaggio. Viene presentato un confronto dei metodi di previsione, con DMC che è il più accurato e vdW-DF2 è la sua corrispondenza più vicina. Credito:Kenta Hongo di JAIST
L'energia a idrogeno ha il potenziale per essere una misura chiave per raggiungere l'obiettivo di emissioni nette di zero delle Nazioni Unite, ma il suo uso industriale è stato ostacolato dalla difficoltà di immagazzinamento e manipolazione. L'idrogeno diventa un gas a una temperatura molto bassa (-252°C), il che rende difficile la sua conservazione a temperatura ambiente. L'interazione tra l'idrogeno e il suo materiale di stoccaggio è semplicemente troppo debole per persistere a temperatura ambiente. Ciò rende la progettazione dei materiali di stoccaggio cruciale per raggiungere l'obiettivo di portare l'energia dell'idrogeno nell'uso quotidiano.
È qui che entra in gioco la progettazione dei materiali computazionali. Durante lo sviluppo della tecnologia dell'idrogeno è possibile risparmiare molto tempo e fatica progettando un materiale su un computer e simulando la sua capacità di stoccaggio dell'idrogeno. Ma le previsioni diventano molto limitate nel loro uso a meno che non siano accurate e possano essere fatte a un costo computazionale ragionevole. In un recente studio pubblicato su ACS Omega , gli scienziati sviluppano un sistema computazionalmente costoso, ma nuovo metodo altamente accurato per prevedere lo stoccaggio dell'idrogeno:"Migliorare l'affidabilità delle previsioni per le simulazioni può aiutare ad accelerare lo sviluppo di materiali per lo stoccaggio del combustibile a idrogeno e portare a una società più efficiente dal punto di vista energetico, " afferma il Dr.Kenta Hongo del Japan Advanced Institute of Science and Technology (JAIST), che ha condotto lo studio.
Una delle forze fondamentali di attrazione tra gli oggetti è la forza di van der Waals, che definisce l'interazione tra atomi o molecole in base alla distanza tra loro. Poiché la forza di Van der Waals è la conseguenza di processi quantistici piuttosto complicati, i trattamenti convenzionali non potrebbero descriverlo bene, e quindi le simulazioni finora sono al livello di stime approssimative di esso. Ma è giusto farlo quando si simula lo stoccaggio dell'idrogeno? Questa era la preoccupazione principale del Dr. Hongo e del suo team.
Per rispondere a questa domanda, hanno esaminato i nanotubi di carburo di silicio, uno dei materiali più promettenti per lo stoccaggio dell'idrogeno. Utilizzando una tecnica computazionale chiamata diffusione Monte Carlo (DMC), hanno creato un modello che rappresentava le forze di van der Waals durante la simulazione dello stoccaggio dell'idrogeno nei nanotubi di carburo di silicio. La maggior parte dei modelli convenzionali considera le interazioni tra idrogeno e nanotubi di carburo di silicio nel loro insieme, ma il metodo DMC utilizza la potenza di un supercomputer per ricostruire fedelmente il meccanismo di interazione seguendo la disposizione dei singoli elettroni. Ciò rende il modello DMC il metodo di previsione più accurato fino ad oggi. Utilizzando il modello DMC, i ricercatori sono stati anche in grado di prevedere quanta energia sarebbe necessaria per rimuovere l'idrogeno dal suo stoccaggio, e quanto era probabile che fosse lontano l'idrogeno dalla superficie del nanotubo di carburo di silicio. Hanno quindi confrontato i risultati della loro modellazione con quelli ottenuti tramite metodi di previsione convenzionali.
I metodi di previsione convenzionali sono generalmente basati su tecniche computazionali chiamate teoria del funzionale della densità (DFT). DFT utilizza funzionali (descrizioni di modelli di interazioni quantistiche) che descrivono le variazioni spaziali della densità elettronica per determinare le proprietà di sistemi complessi. Mentre ci sono stati diversi studi basati su DFT sullo stoccaggio di idrogeno su nanotubi di carburo di silicio, nessuno di loro ha incorporato le forze di van der Waals nelle sue previsioni. I funzionali DFT corretti da Van der Waals hanno, però, stato impiegato nella previsione di altri materiali. Il Dr. Hongo e il team hanno simulato lo stoccaggio dell'idrogeno utilizzando un'ampia gamma di funzionali DFT, quelli con le correzioni di van der Waals e quelli senza. Hanno scoperto che i funzionali DFT senza le correzioni di van der Waals stimavano erroneamente l'energia richiesta per lo stoccaggio dell'idrogeno del 4-14%. D'altra parte, I funzionali DFT corretti da van der Waals hanno prodotto risultati abbastanza simili a quelli di DMC. Inoltre, hanno scoperto che il contributo della forza di van der Waals all'energia di accumulo era di circa il 9-29%, che non è affatto insignificante.
Questi risultati, Il dottor Hongo crede, può essere un trampolino di lancio per ulteriori innovazioni nella tecnologia di simulazione dello stoccaggio dell'idrogeno. "Sebbene il metodo DMC sia computazionalmente costoso, può essere utilizzato per chiarire le peculiarità (tendenze di errore di previsione) di ciascun metodo di previsione. Questo ci aiuterà a capire di quale previsione fidarci, e anche come modificare i metodi di previsione per renderli più utili, " lui spiega.