Astratto grafico. Credito:Journal of Molecular Biology (2022). DOI:10.1016/j.jmb.2022.167525
Quali geni sono specifici per un certo tipo di cellula, cioè "marcano" la loro identità? Con l'aumento delle dimensioni dei set di dati al giorno d'oggi, rispondere a questa domanda è spesso difficile. Spesso, i geni marcatori sono semplicemente geni che sono stati trovati in specifiche popolazioni cellulari. Tuttavia, molti più geni potrebbero essere caratteristici di un particolare tipo di cellula ma rimangono da scoprire.
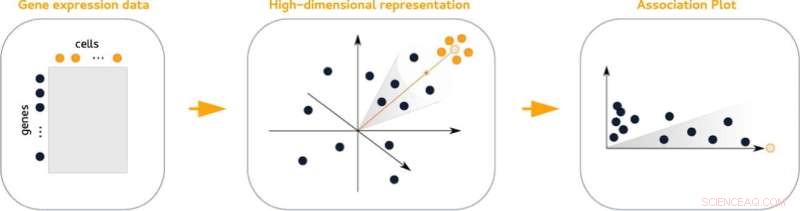
I grafici di associazione (APL), un nuovo metodo statistico per visualizzare l'attività genica all'interno di un cluster di cellule, rendono più facile trovare i suoi geni marcatori. I grafici confrontano l'attività dei geni di un dato cluster con tutti gli altri cluster del set di dati. Inoltre, rendono facile vedere quali geni sono condivisi con altri cluster.
"I grafici di associazione non solo ci consentono di identificare nuovi geni marcatori. Funziona anche al contrario:siamo in grado di abbinare gruppi di identità sconosciuta in un set di dati a tipi di cellule, sulla base di un elenco fornito di geni marcatori", afferma Elzbieta Gralinska del Max Planck Institute for Molecular Genetics di Berlino.
Il biotecnologo lavora nel team di Martin Vingron, che ha sviluppato la tecnica. I ricercatori hanno dimostrato la funzionalità della tecnica su due set di dati pubblicamente disponibili e hanno pubblicato i risultati nel Journal of Molecular Biology . Inoltre, APL è stato rilasciato come modulo gratuito per l'ambiente statistico R. Il pacchetto APL consente ai ricercatori di ispezionare visivamente i propri dati unicellulari e selezionare i singoli geni con il cursore per apprendere dettagli più approfonditi.
Analisi e raggruppamento di singole celle
Perché è necessario identificare i geni marcatori in primo luogo? Le moderne tecnologie di sequenziamento sono in grado di decifrare singole molecole di RNA in singole cellule. Da un campione di sangue, ad esempio, è possibile separare ogni cellula e decodificare un campione di RNA della cellula. Questi dati unicellulari rappresentano i geni attivi che sono stati trascritti in molecole di RNA.
Il vantaggio:invece di interrogarsi su quale tipo di cellula appartenga un particolare RNA, può essere fatto risalire alla sua cellula di origine. Lo svantaggio:il sequenziamento di migliaia di RNA in ogni singola cellula su decine di migliaia di cellule produce quantità straordinarie di dati.
Una via d'uscita è ordinare le cellule in base al loro contenuto di RNA. "I dati a cellula singola sono composti da un mix selvaggio di molti tipi cellulari diversi. Siamo interessati a cellule dello stesso tipo di cellula, che dovrebbero comportarsi tutte in modo simile", spiega Martin Vingron. Quindi, ha senso raggruppare celle simili in modo computazionale, dice. "Per noi, i geni marcatori definiscono un tipo di cellula."
Esplorazione interattiva dei cluster di cellule
Utilizzando i dati pubblicamente disponibili dei globuli bianchi, il team ha dimostrato come funziona il nuovo algoritmo. I molti diversi tipi di globuli bianchi come i linfociti T, i linfociti B oi monociti sono tutti raggruppati in gruppi separati. I ricercatori hanno confermato i geni marcatori noti e sono stati in grado di dimostrare che anche i parenti stretti tra le cellule del sangue condividono una grande somiglianza nella loro attività genica.
"Ciascuno dei geni marcatori che abbiamo trovato con APL potrebbe essere stato scoperto da almeno un altro metodo esistente per l'identificazione dei geni marcatori", afferma Gralinska. Ma il vantaggio di APL rispetto agli algoritmi esistenti è la sua rappresentazione grafica dei risultati, dice. "Gli strumenti esistenti forniscono lunghi elenchi di geni e valori di punteggio. Spesso, gli utenti esaminano l'elenco e si fermano a un limite arbitrario".
Al contrario, il nuovo metodo fornisce un modo per visualizzare questi geni, fare clic su ciascuno e dare un'occhiata più da vicino alla sua attività, dice. "Non stiamo solo fornendo elenchi di geni marcatori, stiamo consentendo agli utenti di rivedere il comportamento di questi geni", afferma il ricercatore. "Con i grafici di associazione, possono approfondire i propri dati per saperne di più su ciascun tipo di cellula". Inoltre, dice, è molto facile scomporre il ruolo biologico dei geni più interessanti in un passaggio successivo tramite l'analisi dell'arricchimento dei termini Gene Ontology, che è compatibile con il software APL, qualcosa che considera "una caratteristica molto utile".
Il modello matematico sottostante
I dati ad alta dimensione che contengono informazioni sull'attività attraverso i geni non possono essere rappresentati visivamente senza perdita di informazioni. Lo stesso vale per i dati raggruppati, che complicano l'analisi. "Il nostro trucco è che prendiamo in considerazione molte più di due o tre dimensioni, ma alla fine creiamo un diagramma bidimensionale", afferma Gralinska.
I grafici di associazione sono derivati da una tecnica matematica che incorpora simultaneamente sia i geni che le cellule in uno spazio comune ad alta dimensione. La misurazione delle distanze tra i geni e un determinato cluster di cellule in questo spazio produce coppie di valori che riflettono l'associazione di un gene a un determinato cluster e forniscono informazioni sulla sua associazione ad altri cluster.
"Una lacuna dell'APL è che ci affidiamo a dati pre-cluster, il che significa che dobbiamo fare affidamento su altre tecniche per il clustering", afferma Martin Vingron. "Tuttavia, speriamo che il nostro nuovo metodo trovi molti nuovi utenti. Scopriamo che un processo visivo e interattivo rende semplicemente un'analisi migliore".