Credito:Università della California, San Francisco
La scoperta di farmaci può far venire in mente immagini di camici bianchi e pipette, ma quando Henry Lin, dottorato di ricerca, recentemente deciso di trovare un oppioide migliore con meno effetti collaterali, il suo primo passo è stato quello di accendere i computer.
Utilizzando un programma chiamato DOCK, ha caricato una struttura cristallina del recettore oppioide trovato nel cervello e ha avuto accesso a una libreria virtuale di 3 milioni di composti che potrebbero legarsi a una "tasca" chimica sul recettore. La maggior parte dei farmaci, dagli antibiotici agli antidepressivi, agisce legandosi a siti specifici sulle proteine, ma per essere efficace, devono adattarsi perfettamente.
Il programma ruotava ogni composto intorno, considerata la flessibilità delle sue varie appendici, e dopo aver testato una media di 1,3 milioni di configurazioni per composto, le abbiamo classificate in base al loro potenziale di legame. Il processo, in esecuzione su computer collegati a potenti processori, ci sono voluti circa due settimane.
Studente laureato all'epoca, Lin ha lavorato con il suo consigliere Brian Shoichet, dottorato di ricerca, professore di chimica farmaceutica presso la UC San Francisco School of Pharmacy, e Aashish Manglik, dottorato di ricerca, della Stanford University per spulciare tra i primi 2, 500 composti per fattori aggiuntivi e 23 selezionati per test sperimentali in cellule viventi:camici da laboratorio e pipette.
Sempre più, i ricercatori si stanno rivolgendo a esperimenti virtuali per le fasi iniziali dello sviluppo di farmaci. Con computer sempre più veloci, la fase iniziale e in gran parte di tentativi ed errori dello sviluppo di farmaci può essere ridotta a una questione di giorni, e con librerie online di composti in continua espansione, gli schermi dei farmaci possono comprendere, letteralmente, tutta la chimica conosciuta nel mondo.
Punti di forza e limiti
I ricercatori sono cauti sul potenziale della scoperta di farmaci computazionali - solo una piccola frazione di composti promettenti funziona effettivamente quando testati nella vita reale - ma dicono che uno dei suoi punti di forza è nel rivelare composti completamente nuovi come candidati farmaci.
Shoichet è specializzato in un popolare metodo computazionale noto come docking molecolare. "Il punto in cui si inserisce l'attracco è nelle prime ricerche di scoperta, nel trovare nuove partenze, " Egli ha detto.
La ricerca del suo team per il nuovo oppioide illustra sia i punti di forza che i limiti della scoperta computazionale di farmaci.
Infatti, i candidati oppioidi iniziali identificati attraverso il docking molecolare si sono esibiti solo modestamente nei test sperimentali. "Ancora, l'attività che avevano era altamente riproducibile e le molecole erano altamente nuove, presagio di una nuova biologia, " disse Shoichet.
Il team ha agganciato un altro giro di mescole con strutture simili e ha testato i migliori marcatori. Con i collaboratori della University of North Carolina, Chapel Hill e la Friedrich Alexander University in Germania, hanno identificato il composto più potente e ne hanno ottimizzato la farmacologia con l'elaborazione sintetica guidata dal computer.
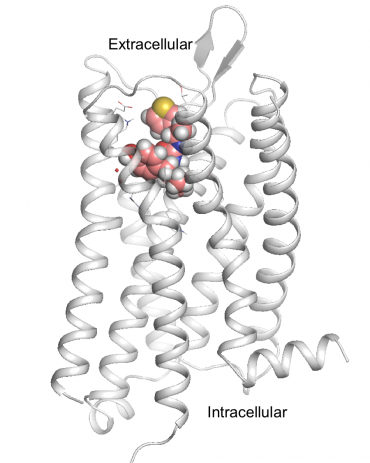
PZM21, il nuovo, candidato farmaco oppioide più sicuro, è mostrato agganciato al recettore della morfina del cervello, il recettore mu-oppioide. Attestazione:Anat Levit
Quel composto vincente, denominato PZM21, è chimicamente diverso da qualsiasi altro attualmente in uso e potrebbe non essere stato trovato con metodi più tradizionali. È un composto completamente progettato in modo computazionale che è più potente della morfina. Nei topi, bloccava efficacemente il dolore senza i soliti effetti collaterali della soppressione respiratoria e della stitichezza e sembrava persino creare meno dipendenza.
L'attracco non è un proiettile d'argento, ma è diventato un potente punto di partenza per il lungo, processo interdisciplinare di sviluppo del farmaco. Tra i suoi maggiori contributi ci sono stati gli inibitori della proteasi che hanno contribuito a rendere l'HIV una malattia curabile. I ricercatori stanno anche utilizzando l'attracco per lo screening dei farmaci candidati per il trattamento del cancro al seno, epatite C, ipertensione, Stafilococco, il virus della SARS e l'influenza.
Tecnologia pionieristica alla UCSF
Il docking molecolare è stato sperimentato tre decenni fa da un giovane chimico fisico della UCSF di nome Tack Kuntz, dottorato di ricerca, ora professore emerito alla Scuola di Farmacia. Quando Kuntz arrivò al campus nei primi anni '70, prevaleva ancora l'approccio tradizionale alla scoperta di farmaci.
Come lo descrisse Kuntz, il processo si basava sul caso e su pochissima teoria:"Esci e trovi nuovi composti naturali e li riporti per testare in un laboratorio. Basta mettere insieme sostanze chimiche con un organismo e vedere cosa succede".
I chimici farmaceutici non si sono mai preoccupati dei dettagli molecolari di come i farmaci interagissero con il corpo. Molti farmaci, compresi i primi antibiotici, era stato scoperto per caso, ma Kuntz, dopo aver visto la nuova comprensione molecolare spazzare il campo della biologia, sentiva che era giunto il momento per un aggiornamento simile in farmacologia.
"La visione della biologia basata sull'obiettivo - che è possibile comprendere la biologia attraverso proteine indipendenti e prodotti genici - aveva già preso il sopravvento, ma la farmacologia era indietro di un decennio, " disse Shoichet, che era uno studente laureato nel laboratorio di Kuntz negli anni '80.
Kuntz e i suoi colleghi hanno iniziato a lavorare verso un approccio più razionale alla progettazione di farmaci in cui hanno cercato di identificare composti che potessero adattarsi a recettori specifici sulle proteine, come trovare il pezzo mancante di un puzzle. Nel 1982, hanno pubblicato un documento che descrive il primo programma di docking molecolare che potrebbe "esplorare allineamenti geometricamente fattibili di ligandi e recettori di struttura nota".
Kuntz ha inviato 10, 000 copie di quel primo programma di attracco ai ricercatori di tutto il paese. Prossimamente, altri ricercatori stavano sviluppando programmi di calcolo simili e l'entusiasmo si diffuse rapidamente al di fuori del mondo accademico. Entro gli anni '90, ogni grande azienda farmaceutica aveva aperto un'unità di scoperta di farmaci computazionale.
Recuperare un'idea
Nonostante l'entusiasmo iniziale, però, la scoperta computazionale di farmaci non ha portato a risultati rapidi. L'idea di Kuntz era arrivata in anticipo sui tempi. Ci vorrebbero decenni di progressi incrementali nella biologia molecolare, tecnologia di imaging e informatica, prima che la scoperta computazionale di farmaci possa iniziare a mantenere la sua promessa.

Tack Kuntz, dottorato di ricerca, e i suoi colleghi nel 1982 hanno pubblicato un articolo che descrive il primo programma di docking molecolare che potrebbe "esplorare allineamenti geometricamente fattibili di ligandi e recettori di struttura nota". Credito:Università della California, San Francisco
Una delle principali limitazioni negli anni '90 era la mancanza di strutture proteiche note. Senza questi, c'erano pochi bersagli per cui trovare la droga. Nei decenni successivi, migliaia di strutture proteiche di possibili bersagli farmacologici sono state rivelate dalla cristallografia a raggi X e dalla risonanza magnetica nucleare.
La scoperta del nuovo candidato agli oppiacei, ad esempio, è stato possibile solo grazie alle strutture cristalline recentemente determinate dei recettori accoppiati a proteine G, una famiglia di proteine che include il recettore degli oppioidi.
Anche le librerie virtuali di composti sono cresciute in modo esponenziale. Nel 1991, un database potrebbe contenere 55, 000 composti; ora contengono decine di milioni. "La portata della chimica che stiamo campionando è aumentata più o meno alla stessa velocità della legge di Moore, "Shoichet ha detto. "C'è una fame insaziabile di sempre più molecole."
I programmi di docking odierni sono in grado di modellare realisticamente le interazioni a livello atomico tra un farmaco e il suo bersaglio, ma alcuni dettagli complicati, come il modo in cui le forze atomiche cambiano quando una molecola di farmaco sposta l'acqua nel sito di legame, rimangono sfide in corso nel campo.
Promesse e prove
L'aggancio molecolare non è l'unica forma di progettazione di farmaci basata su computer. Presso l'Istituto UCSF per le scienze della salute computazionale (ICHS), dozzine di ricercatori stanno esplorando una miriade di metodi computazionali per far progredire la ricerca medica.
Michael Keiser, dottorato di ricerca, un membro dell'ICHS e un assistente professore presso l'Istituto di malattie neurodegenerative, sta studiando farmaci che colpiscono più bersagli molecolari contemporaneamente, come se suonasse un accordo piuttosto che una singola nota. Questa azione multi-target è stata a lungo considerata la causa di effetti collaterali indesiderati, ma può anche essere indirizzato al trattamento di malattie complesse.
Solo nei primi anni 2000 i ricercatori sono arrivati a riconoscere che molti farmaci esistenti agiscono attraverso più di un obiettivo:antipsicotici, Per esempio, che colpiscono i recettori della serotonina e della dopamina. Ora stanno intenzionalmente progettando farmaci per farlo.
"Per alcune malattie che non hanno ancora cure, forse è perché non c'è una singola proteina che devi attivare o disattivare; e se invece il farmaco dovesse colpire più bersagli?" ha detto Keiser, che era uno studente laureato di Shoichet.
Nel suo laboratorio, Keizer utilizza metodi computazionali per identificare modelli chimici tra farmaci che si legano allo stesso insieme di bersagli e trovare nuovi composti con farmacologia corrispondente. Questo approccio computazionale può riconoscere somiglianze tra i composti che le analisi più convenzionali mancherebbero. Keizer sta ora guardando verso la tecnologia dell'intelligenza artificiale, noto come apprendimento profondo, per un riconoscimento ancora migliore dei modelli.
Anche quando i metodi di calcolo decollano, la loro prova è ancora nel mondo reale, nelle cellule, modelli animali, e infine in clinica. "Per un po' era comune pubblicare articoli con previsioni sulle attività di una piccola molecola, ma nessun test effettivo di queste previsioni, perché gli esperimenti per farlo erano costosi, difficile o esoterico, " ha detto Keiser.
Poiché la necessità di collaborazione è diventata chiara, la partnership tra la previsione computazionale e gli esperimenti di laboratorio umido si è notevolmente rafforzata nell'ultimo decennio, disse Keiser. "Dopotutto, come puoi migliorare le tue previsioni se non sei sicuro di quali siano sbagliate?"