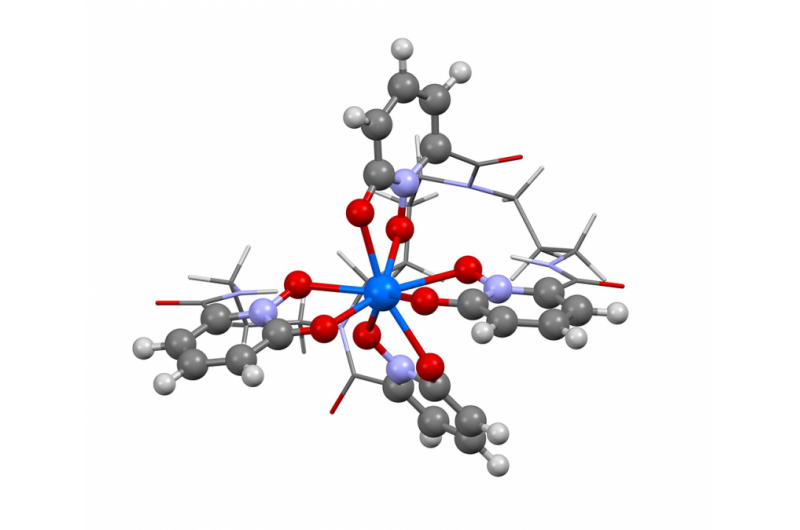
Questa immagine mostra la struttura del berkelio allo stato di ossidazione +IV. I ricercatori hanno utilizzato il nuovo algoritmo del Berkeley Lab per calcolare lo spettro di assorbimento e confermare ciò che diversi risultati sperimentali suggerivano:che l'elemento berkelio si rompe con i suoi coetanei di elementi pesanti assumendo una carica extra positiva quando è legato a una molecola organica sintetica. Questa proprietà potrebbe aiutare gli scienziati a sviluppare metodi migliori per maneggiare e purificare i materiali nucleari. Credito:Bert de Jong, Berkeley Lab
Gli oggetti fosforescenti sembrano magici quando sei un bambino:possono illuminare una stanza buia senza bisogno di elettricità, batterie o una lampadina. Poi ad un certo punto impari la scienza dietro questo fenomeno. I composti chimici chiamati cromofori si eccitano, o eccitato, quando assorbono la luce visibile. Quando tornano al loro stato normale, l'energia immagazzinata viene rilasciata sotto forma di luce, che percepiamo come un bagliore. Nella scienza dei materiali, i ricercatori si affidano a un fenomeno simile per studiare le strutture dei materiali che verranno infine utilizzati nella catalisi chimica, batterie, applicazioni solari e altro ancora.
Quando una molecola assorbe un fotone, la particella fondamentale della luce, gli elettroni nel sistema molecolare vengono promossi da uno stato di bassa energia (fondamento) a uno stato di energia superiore (eccitato). Queste risposte risuonano a frequenze luminose specifiche, lasciando "impronte spettrali" che illuminano le strutture atomiche ed elettroniche del sistema oggetto di studio.
Negli esperimenti, le "impronte spettrali" o spettro di assorbimento, vengono misurati con strutture all'avanguardia come l'Advanced Light Source (ALS) presso il Lawrence Berkeley National Laboratory (Berkeley Lab) del Dipartimento dell'Energia degli Stati Uniti. Nelle simulazioni al computer, queste misurazioni vengono in genere acquisite con un metodo quantomeccanico chiamato teoria funzionale della densità dipendente dal tempo (TDDFT). I modelli computazionali sono fondamentali per aiutare i ricercatori a ottenere il massimo dai loro esperimenti prevedendo e convalidando i risultati.
Eppure, nonostante la sua utilità, ci sono momenti in cui TDDFT non può non essere utilizzato per calcolare lo spettro di assorbimento di un sistema perché richiederebbe troppo tempo e risorse informatiche. È qui che torna utile una nuova "scorciatoia" matematica sviluppata dai ricercatori della divisione di ricerca computazionale (CRD) del Berkeley Lab. Il loro algoritmo accelera i calcoli di assorbimento di un fattore cinque, quindi le simulazioni che impiegavano da 10 a 15 ore per il calcolo ora possono essere eseguite in circa 2,5 ore.
Un documento che descrive questo metodo è stato pubblicato sul Journal of Chemical Theory and Computation (JCTC). E il nuovo approccio per il calcolo dello spettro di assorbimento sarà incorporato in una prossima versione della suite di software di chimica computazionale NWChem ampiamente utilizzata entro la fine dell'anno.
Nuovi algoritmi portano a risparmi computazionali
Per studiare la struttura chimica di nuove molecole e materiali, gli scienziati in genere sondano il sistema con uno stimolo esterno, in genere un laser, quindi cercano piccoli cambiamenti elettronici. Matematicamente, questo cambiamento elettronico può essere espresso come un problema agli autovalori. Risolvendo questo problema agli autovalori, i ricercatori possono ottenere una buona approssimazione dello spettro di assorbimento, che a sua volta rivela le frequenze di risonanza del sistema in esame. Nel frattempo, l'autovettore corrispondente viene utilizzato per calcolare l'intensità con cui il sistema ha risposto allo stimolo. Questo è essenzialmente il principio alla base dell'approccio TDDFT, che è stato implementato in diversi pacchetti software di chimica quantistica, inclusa la suite software open source NWChem.
Sebbene questo approccio si sia dimostrato vincente, ha limitazioni per i grandi sistemi. Più ampia è la gamma di energia delle risposte elettroniche che un ricercatore cerca di catturare in un sistema, più autovalori e autovettori devono essere calcolati, il che significa anche che sono necessarie più risorse di calcolo. In definitiva, lo spettro di assorbimento di un sistema molecolare con più di 100 atomi diventa proibitivo da calcolare con questo metodo.
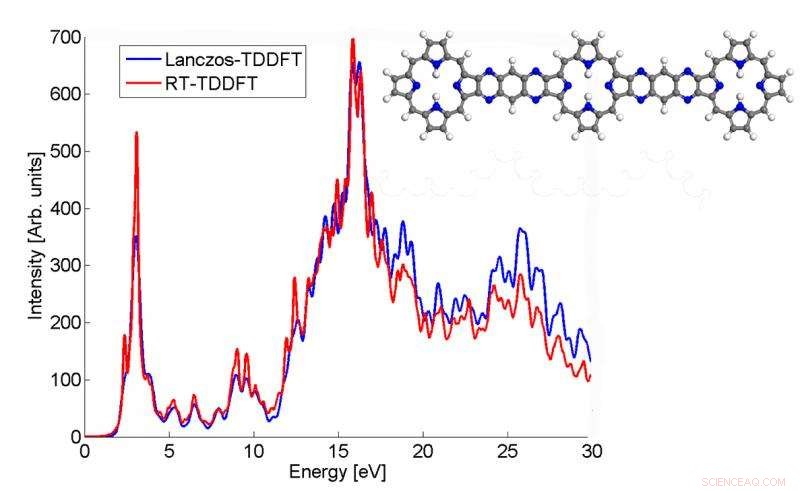
Questo grafico mostra come lo spettro di assorbimento di una molecola p3b2 calcolato dall'algoritmo di Lanczos corrisponda al risultato TDDFT in tempo reale. Credito:Chao Yang, Berkeley Lab
Per superare questi limiti, i matematici del CRD hanno sviluppato una tecnica per calcolare direttamente lo spettro di assorbimento senza calcolare esplicitamente gli autovalori della matrice.
"Tradizionalmente, i ricercatori hanno dovuto calcolare gli autovalori e gli autovettori di matrici molto grandi per generare lo spettro di assorbimento, ma ci siamo resi conto che non è necessario calcolare ogni singolo autovalore per ottenere una visione accurata dello spettro di assorbimento, "dice Chao Yang, un matematico CRD che ha guidato lo sviluppo del nuovo approccio.
Riformulando il problema come approssimazione di una funzione di matrice, avvalendosi di una apposita trasformazione e sfruttando la sottostante simmetria rispetto ad una metrica non euclidea, Yang e i suoi colleghi sono stati in grado di applicare l'algoritmo di Lanczos e un metodo Kernal Polynomial Method (KPM) per approssimare lo spettro di assorbimento di diverse molecole. Entrambi questi algoritmi richiedono una memoria relativamente bassa rispetto alle alternative non simmetriche, che è la chiave per il risparmio computazionale.
Poiché questo metodo richiede meno potenza di calcolo per ottenere un risultato, i ricercatori possono anche calcolare facilmente lo spettro di assorbimento per sistemi molecolari con diverse centinaia di atomi.
"Questo metodo è un significativo passo avanti perché ci consente di modellare lo spettro di assorbimento di sistemi molecolari di centinaia di atomi a un costo computazionale inferiore". dice Niranjan Govind, un chimico computazionale presso il Pacific Northwest National Laboratory che ha collaborato con il team del Berkeley Lab allo sviluppo del metodo nel programma di chimica computazionale NWChem.
Recentemente gli scienziati del Berkeley Lab hanno utilizzato questo metodo per calcolare lo spettro di assorbimento e confermare ciò che diversi risultati sperimentali suggerivano:che l'elemento berkelio rompe la forma con i suoi simili elementi pesanti assumendo una carica positiva extra quando legato a una molecola organica sintetica. Questa proprietà potrebbe aiutare gli scienziati a sviluppare metodi migliori per maneggiare e purificare i materiali nucleari. Un articolo che evidenzia questo risultato è apparso il 10 aprile sulla rivista Chimica della natura .
"I risultati sperimentali suggerivano questo comportamento insolito nel berkelio, ma non c'erano abbastanza prove sperimentali per dire di sì, 100 percento, questo è quello che stiamo vedendo, ", afferma il coautore dello studio Wibe Albert de Jong, uno scienziato CRD. "Per essere sicuri al 100%, abbiamo fatto grandi simulazioni computazionali e le abbiamo confrontate con i dati sperimentali e abbiamo stabilito che erano, infatti, vedere il berkelio in uno stato di ossidazione insolito."
Questo nuovo algoritmo è stato sviluppato attraverso un progetto Scientific Discovery through Advanced Computing (SciDAC) supportato dall'Office of Science del DOE, incentrato sul progresso di software e algoritmi per le reazioni fotochimiche. I progetti SciDAC in genere riuniscono un team interdisciplinare di ricercatori per sviluppare metodi computazionali nuovi e innovativi per affrontare alcuni dei problemi scientifici più impegnativi.
"La natura interdisciplinare di SciDAC è un modo molto efficace per facilitare la scienza rivoluzionaria, poiché ogni membro del team porta una prospettiva diversa alla risoluzione dei problemi, " dice Yang. "In questo ambiente dinamico, matematici, come me, collaborare con scienziati del dominio per identificare i colli di bottiglia computazionali, quindi utilizziamo tecniche matematiche all'avanguardia per affrontare e superare queste sfide".