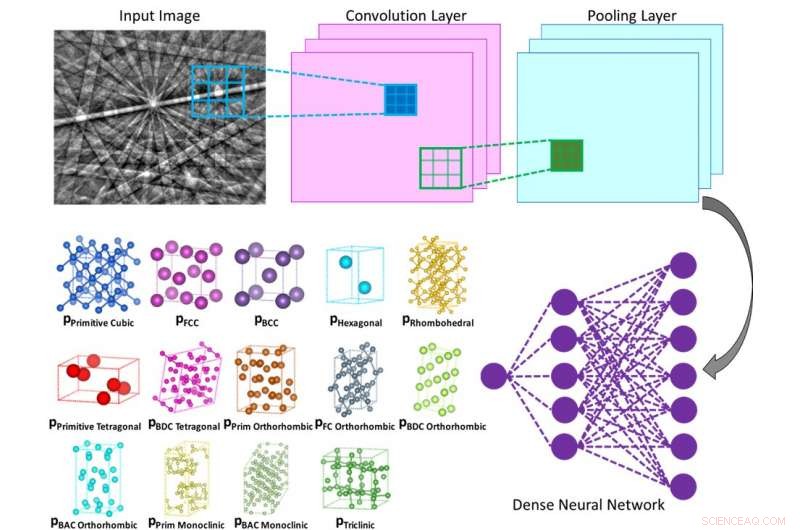
Illustrazione del funzionamento interno di una rete neurale convoluzionale che calcola la probabilità che il modello di diffrazione in ingresso appartenga a una data classe (ad esempio reticolo di Bravais o gruppo spaziale). Crediti:Vecchio lab/Science
I nanoingegneri dell'Università della California a San Diego hanno sviluppato un metodo basato su computer che potrebbe rendere meno laborioso determinare le strutture cristalline di vari materiali e molecole, comprese le leghe, proteine e farmaci. Il metodo utilizza un algoritmo di apprendimento automatico, simile al tipo utilizzato per il riconoscimento facciale e le auto a guida autonoma, analizzare in modo indipendente i modelli di diffrazione elettronica, e farlo con almeno il 95% di precisione.
Il lavoro è pubblicato nel numero del 31 gennaio di Scienza .
Un team guidato dal professore di nanoingegneria della UC San Diego Kenneth Vecchio e dal suo dottorato di ricerca. studente Kevin Kaufmann, chi è il primo autore dell'articolo, sviluppato il nuovo approccio. Il loro metodo prevede l'utilizzo di un microscopio elettronico a scansione (SEM) per raccogliere modelli di diffrazione a retrodiffusione di elettroni (EBSD). Rispetto ad altre tecniche di diffrazione elettronica, come quelli in microscopia elettronica a trasmissione (TEM), L'EBSD basato su SEM può essere eseguito su campioni di grandi dimensioni e analizzato su più scale di lunghezza. Ciò fornisce informazioni locali sub-micron mappate su scale centimetriche. Per esempio, un moderno sistema EBSD consente la determinazione di strutture a grana fine, orientamenti dei cristalli, tensione o deformazione residua relativa, e altre informazioni in una singola scansione del campione.
Però, lo svantaggio dei sistemi EBSD commerciali è l'incapacità del software di determinare la struttura atomica dei reticoli cristallini presenti all'interno del materiale in analisi. Ciò significa che un utente del software commerciale deve selezionare fino a cinque strutture cristalline che si presume siano presenti nel campione e quindi il software tenta di trovare probabili corrispondenze con il modello di diffrazione. La natura complessa del modello di diffrazione spesso fa sì che il software trovi false corrispondenze di strutture nell'elenco selezionato dall'utente. Di conseguenza, l'accuratezza della determinazione del tipo di reticolo da parte del software esistente dipende dall'esperienza dell'operatore e dalla precedente conoscenza del proprio campione.
Il metodo che il team di Vecchio ha sviluppato fa tutto questo in autonomia, poiché la rete neurale profonda analizza in modo indipendente ogni modello di diffrazione per determinare il reticolo cristallino, tra tutti i possibili tipi di struttura reticolare, con un alto grado di precisione (superiore al 95%).
Una vasta gamma di aree di ricerca tra cui farmacologia, biologia strutturale, e la geologia dovrebbero trarre vantaggio dall'utilizzo di algoritmi automatizzati simili per ridurre la quantità di tempo necessaria per l'identificazione strutturale dei cristalli, ricercatori hanno detto.