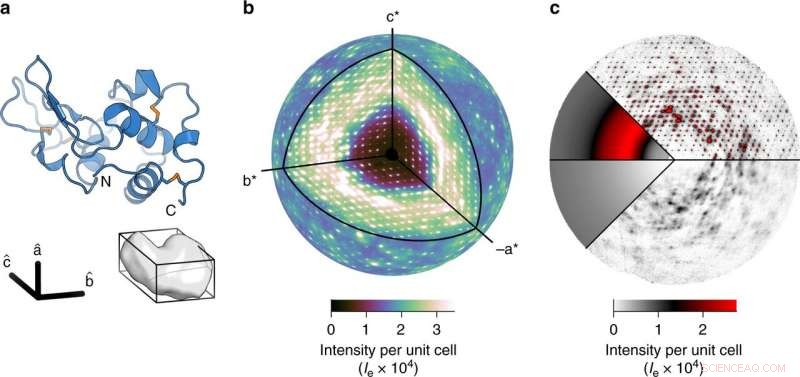
un diagramma a nastro del lisozima (in alto) e della cellula unitaria triclina contenente una proteina (in basso). b È stata ottenuta una mappa tridimensionale altamente dettagliata della diffusione diffusa. La sfera esterna viene disegnata con una risoluzione di 2 Å. c Lo scattering totale è costituito da tre componenti:scattering anelastico Compton (in basso a sinistra), un ampio anello isotropo che domina il segnale diffuso (in alto a sinistra), e caratteristiche variazionali nella diffusione diffusa (a destra). Aloni intensi sono visibili negli strati contenenti picchi di Bragg (l = 0 piano, In alto a destra). Lo scattering nuvoloso si visualizza meglio nei piani a metà strada tra i picchi di Bragg (l = 1∕2 piano, in basso a destra). Credito: Comunicazioni sulla natura (2020). DOI:10.1038/s41467-020-14933-6
I biologi strutturali di Cornell hanno adottato un nuovo approccio all'utilizzo di un metodo classico di analisi a raggi X per catturare qualcosa di cui il metodo convenzionale non aveva mai tenuto conto:il movimento collettivo delle proteine. E lo hanno fatto creando un software per unire faticosamente insieme i frammenti di dati che di solito vengono ignorati nel processo.
La loro carta, "Dispersione diffusa di raggi X da movimenti correlati in un cristallo proteico, " pubblicato il 9 marzo in Comunicazioni sulla natura .
Come biologo strutturale, Nozomi Ando, SM. '04, dottorato di ricerca '08, professore assistente di chimica e biologia chimica, è interessato a tracciare il movimento delle proteine, e le loro parti interne, per comprendere meglio la funzione delle proteine. Questo tipo di movimento è ben noto ma è stato difficile da documentare perché la tecnica standard per l'imaging delle proteine è la cristallografia a raggi X, che produce essenzialmente istantanee statiche.
"Perché stiamo studiando sistemi biologici davvero impegnativi, il gruppo deve spesso sperimentare anche nuovi metodi strutturali, " ha detto il ricercatore post-dottorato Steve Meisburger, dottorato di ricerca '14, l'autore principale del documento. "Una delle domande a cui siamo stati interessati fin dall'inizio è come i sottili movimenti respiratori di una proteina dirigano la funzione biochimica".
I ricercatori hanno portato il loro progetto alla Cornell High Energy Synchrotron Source (CHESS), dove hanno approfittato del rivelatore Pilatus 6M pixel-array della struttura, che ha permesso loro di realizzare immagini ad altissima risoluzione.
Per questo lavoro, come nella normale cristallografia, I raggi X sono stati trasmessi a un cristallo campione. Il rivelatore di pixel-array ha registrato l'intensità dei raggi X che sono stati diffratti dalle proteine del cristallo, codificando così la struttura atomica. Qualsiasi disturbo, cioè movimento:all'interno del cristallo faceva rimbalzare altri fotoni, creando un segnale di fondo molto debole chiamato diffusione diffusa. Queste informazioni sono state tradizionalmente scartate durante l'elaborazione dei dati.
"I fotoni vanno ovunque, e il segnale appare estremamente debole perché è diffuso, " disse Ando, l'autore senior del documento. "Per decenni, le persone non potevano misurarlo con precisione, e non sapevano come interpretarlo."
Meisburger ha creato un software per elaborare i circa 50 milioni di punti dati unici, ottenendo una mappa tridimensionale di alta qualità. Con grande sorpresa dei ricercatori, la mappa ha rivelato che una componente significativa di questo modello di dispersione diffusa era in realtà il risultato della vibrazione del reticolo proteico. Questo movimento oscillante era così dominante, sembrava oscurare qualsiasi movimento all'interno delle proteine, che inizialmente è stata una delusione per i ricercatori.
Ma dopo aver tenuto conto di queste vibrazioni reticolari nelle simulazioni, i ricercatori hanno identificato anche i movimenti interni delle proteine. Questi movimenti includevano l'apertura e la chiusura del sito attivo della proteina.
"Immagina il cristallo come una fila di persone che cercano di camminare insieme tenendosi per mano, ma allo stesso tempo, ogni individuo potrebbe fare qualcosa di leggermente diverso, " disse Ando. "Il segnale di tutti che si muovono insieme è dominante, quindi non potevamo discernere il sottile segnale che proveniva dagli individui. Era qualcosa di cui non si era mai tenuto conto".
Questo nuovo approccio alla diffusione diffusa potrebbe aiutare i ricercatori a ottenere un quadro più chiaro della struttura e della dinamica delle proteine e, in definitiva, una migliore comprensione delle reazioni biochimiche.
"Vogliamo davvero spingerlo in una direzione in cui molte persone possono usare la tecnica e imparare qualcosa di nuovo sulle loro proteine, " Meisburger ha detto. "Una cosa grandiosa è che ottieni la diffusione diffusa gratuitamente ogni volta che fai un normale esperimento di cristallografia. Questa tecnica aggiunge davvero informazioni a ciò che normalmente otterresti."