Ammoniaca (NH3 ) è una molecola importante con molte applicazioni. Prodotto finale del famoso processo Haber-Bosch, viene comunemente sintetizzato per catturare l'azoto per i fertilizzanti e viene utilizzato per la refrigerazione, nei prodotti per la pulizia e nella produzione di prodotti farmaceutici. Recentemente, questa modesta molecola ha attirato l'interesse anche come potenziale risorsa per affrontare una delle sfide più urgenti di oggi:la necessità di combustibili rinnovabili affidabili e abbondanti.
L'ammoniaca è stabile e sicura da maneggiare, è combustibile e contiene la più grande frazione di idrogeno di qualsiasi molecola ad eccezione dell'idrogeno puro stesso. Questi fattori promettono di renderlo un’alternativa fattibile ai vettori energetici basati sul carbonio che stanno guidando il cambiamento climatico. La ricerca ha iniziato a esplorare come l’ammoniaca potrebbe essere utilizzata per alimentare direttamente motori, turbine a gas e celle a combustibile a idrogeno, ad esempio. Si ritiene inoltre che l'ammoniaca potrebbe essere utilizzata per immagazzinare energia per i periodi in cui altre energie rinnovabili come l'energia eolica e solare non possono soddisfare la domanda.
Si sa molto sull’ammoniaca, ma questo interesse nell’usarla come combustibile ha avviato la ricerca di nuove tecnologie per l’ammoniaca. Ciò, a sua volta, ha portato ad una maggiore necessità tra gli ingegneri chimici di dati accurati che descrivano le proprietà termodinamiche fondamentali dell'ammoniaca. Tali proprietà includono un’ampia varietà di tratti misurabili come gli equilibri di fase, la densità o la capacità termica, ad esempio, che caratterizzano i sistemi fisici e determinano il funzionamento dei processi chimici. Nel caso dell'ammoniaca, gli ingegneri vorrebbero anche avere una migliore conoscenza di come tali proprietà cambiano quando si mescola l'ammoniaca con altre molecole. Tale conoscenza potrebbe aiutarli a ottimizzare i processi e le condizioni operative.
Il dottor Jadran Vrabec, attualmente direttore dell'Istituto per le scienze dei processi presso l'Università tecnica di Berlino, ha trascorso gran parte della sua carriera utilizzando il calcolo ad alte prestazioni (HPC) per studiare le proprietà termodinamiche a livello molecolare. "Le proprietà termodinamiche sono determinate al 100% dalle interazioni molecolari", spiega. "E poiché queste interazioni avvengono così velocemente e su scala così piccola, è possibile studiarle solo eseguendo simulazioni su larga scala utilizzando supercomputer."
In un recente articolo pubblicato sul Journal of Chemical &Engineering Data , lui e il coautore Erich Mace della TU di Berlino riferiscono sui risultati di simulazioni focalizzate sulle proprietà termodinamiche di miscele contenenti ammoniaca. Prodotti utilizzando il supercomputer Hawk presso il Centro di calcolo ad alte prestazioni di Stoccarda (HLRS), i loro risultati aggiungono dati preziosi che potrebbero supportare lo sviluppo di nuove applicazioni dell'ammoniaca. I risultati potrebbero anche aiutare a valutare l'accuratezza di altri dati esistenti, garantendo che gli ingegneri dispongano delle migliori informazioni disponibili per lavorare con la sostanza.
Le simulazioni su larga scala forniscono informazioni uniche sulle proprietà termodinamiche
Vrabec utilizza da molto tempo le risorse di supercalcolo HLRS per la dinamica molecolare e le simulazioni Monte Carlo. Il suo approccio si basa sui concetti della termodinamica che furono articolati per la prima volta da Ludwig Boltzmann nel XIX secolo, ma divennero pratici da applicare solo negli anni '50 con l'arrivo dei primi computer. Da allora, il campo è avanzato parallelamente allo sviluppo di supercomputer più grandi e veloci, al punto che le simulazioni di Vrabec ora tracciano simultaneamente i movimenti individuali e le interazioni di miliardi o addirittura trilioni di molecole. Utilizzando il software sviluppato dal suo laboratorio per acquisire selettivamente i dati di interesse, può quindi studiare le proprietà termodinamiche delle molecole.
Vrabec utilizza due codici di simulazione chiamati ms2 e ls1, che ha sviluppato e ottimizzato nel corso di una lunga e fruttuosa collaborazione con i membri dello staff HLRS Martin Bernreuther e Christoph Niethammer. Nel 2019 il team ha addirittura stabilito un record mondiale per il più grande sistema molecolare mai simulato utilizzando metodi di dinamica molecolare. Utilizzando ls1, hanno adattato in modo efficiente il loro codice a un sistema di 21 trilioni di atomi in cui è stato possibile monitorare ogni singola molecola e le sue interazioni con altre molecole.
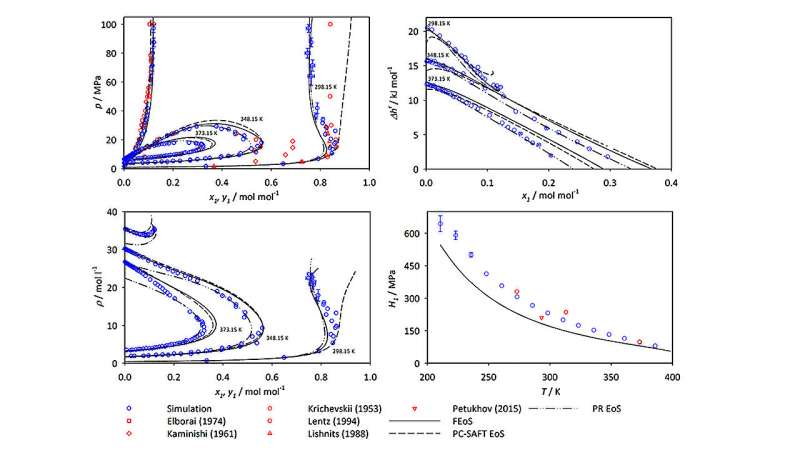
Nel recente lavoro sull'ammoniaca, Mace e Vrabec hanno eseguito simulazioni di dinamica molecolare e Monte Carlo utilizzando ms2 per studiare cinque miscele comunemente usate che coinvolgono l'ammoniaca nei processi di ingegneria chimica:argon-ammoniaca, metano-ammoniaca, idrogeno-ammoniaca, azoto-ammoniaca e ossigeno -ammoniaca. Per ciascuna miscela le simulazioni hanno generato dati che descrivono l'equilibrio vapore-liquido (VLE), una misura della distribuzione delle molecole in un sistema attraverso le fasi vapore o liquida, per un'ampia gamma di temperature e pressioni.
Nel loro articolo, Mace e Vrabec sottolineano che i dati VLE vengono spesso utilizzati nello sviluppo di equazioni di stato per i fluidi industriali; ovvero, i dati possono essere utilizzati per prevedere lo stato della materia in diverse condizioni fisiche dovute a cambiamenti di temperatura, pressione, volume o composizione. Tali informazioni sono essenziali per determinare miscele e condizioni di lavoro ottimali nelle applicazioni industriali.
Le simulazioni molecolari di Vrabec sono particolarmente preziose perché possono essere utilizzate per studiare una gamma di scale molto più ampia di quanto sia possibile utilizzando approcci sperimentali.
"Nelle nostre simulazioni, abbiamo fornito misurazioni delle proprietà termodinamiche anche fino a pressioni di 50 megapascal. Si tratta di 500 volte la nostra pressione dell'aria ambiente", osserva Vrabec. "Sebbene i dati sulle miscele di ammoniaca siano stati raccolti per più di un secolo, la copertura dei dati è sorprendentemente ristretta. La ragione è che lo sforzo per misurarli sperimentalmente è proibitivamente enorme. Richiederebbe costose attrezzature speciali che sarebbero pericolose da utilizzare. In simulazioni al computer, possiamo ottenere risultati in modo sicuro e relativamente economico." I suoi metodi forniscono inoltre un livello di precisione paragonabile a quello degli approcci sperimentali negli intervalli in cui sono disponibili dati sperimentali.
Dati migliori per la ricerca sull'ammoniaca
Quando Mace e Vrabec hanno analizzato i dati della simulazione, hanno dimostrato che, sebbene l’ammoniaca sia un componente di tutti e cinque i sistemi studiati, i grafici risultanti dei valori VLE appaiono notevolmente diversi per le diverse miscele molecolari. Secondo Vrabec, "Il comportamento di fase delle diverse miscele è fortemente determinato dalle interazioni tra le molecole nel sistema. È necessario comprendere queste proprietà se si è interessati a lavorare con miscele di ammoniaca."
L'articolo e i suoi dati supplementari offrono più di 400 nuovi dati per ciascuna miscela studiata. Utilizzando Hawk, sono stati in grado di produrre i risultati di ciascuna miscela in pochi giorni di tempo di calcolo. I risultati saranno di particolare valore per condizioni estreme e difficili da studiare per le quali sono disponibili pochi dati e potrebbero aiutare gli ingegneri a identificare i punti ottimali in cui le condizioni sarebbero ottimali per un trattamento efficiente dell'ammoniaca.
Lo studio ha incluso sia nuovi dati di simulazione sia dati precedentemente pubblicati provenienti dalla letteratura scientifica, consentendo a Mace e Vrabec di confrontare i loro risultati con altri set di dati esistenti di valori VLE. Nella maggior parte dei casi, i risultati corrispondevano strettamente a quelli degli studi precedenti. In alcuni casi, tuttavia, hanno identificato divergenze significative tra i loro risultati e le misurazioni e le previsioni generate sperimentalmente da altri gruppi di ricerca. Gli autori attribuiscono queste discrepanze a limitazioni o imprecisioni nei corrispondenti metodi sperimentali. Suggeriscono inoltre che specifiche fonti di dati sperimentali dovrebbero essere utilizzate con cautela nelle future applicazioni di ricerca o di ingegneria chimica.
Vrabec afferma che nel lavoro recente si è concentrato principalmente sulla simulazione delle proprietà termodinamiche dei sistemi molecolari, generalmente su scala submicrometrica. Nonostante i molti ordini di grandezza che si trovano tra questa scala e il livello dei processi osservabili, esistono metodi accurati per tradurre queste intuizioni a livello molecolare in utili previsioni del mondo reale.
Tuttavia, man mano che i supercomputer diventano più grandi, egli prevede che potrebbe anche diventare possibile simulare non solo le proprietà ma anche i processi termodinamici utilizzando condizioni al contorno vicine alle applicazioni del mondo reale. L'aumento delle prestazioni HPC potrebbe produrre risultati più accurati sui fenomeni dinamici con un migliore rapporto segnale-rumore.
Nel frattempo, però, i risultati del suo team dimostrano il valore della dinamica molecolare e della simulazione Monte Carlo utilizzando il calcolo ad alte prestazioni e forniranno una nuova comprensione del comportamento di fase che gli ingegneri potranno utilizzare per sviluppare nuove tecnologie basate sull'ammoniaca.
Ulteriori informazioni: Erich J. Mace et al, Equilibri della fase fluida ad alta pressione e costanti di Henry dei gas supercritici nell'ammoniaca, Journal of Chemical &Engineering Data (2023). DOI:10.1021/acs.jced.3c00327
Fornito dal Gauss Center for Supercomputing