Un gruppo di ricerca dell’Istituto di chimica organica e biochimica dell’Accademia ceca delle scienze/IOCB di Praga ha sviluppato un nuovo metodo computazionale in grado di descrivere accuratamente come le proteine interagiscono con le molecole di potenziali farmaci e può farlo in poche decine di minuti. Questa nuova funzione di punteggio quantomeccanico può quindi accelerare notevolmente la ricerca di nuovi farmaci. La ricerca è stata pubblicata sulla rivista Nature Communications .
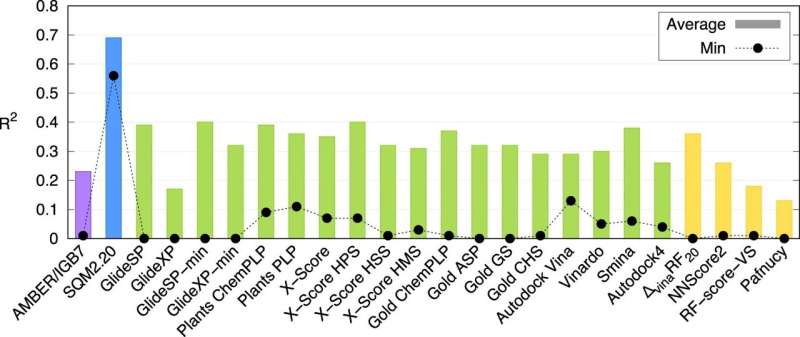
Lo studio dimostra che questo è il primo metodo universalmente applicabile nel suo genere. Gli esperti computazionali dell'IOCB di Praga lo hanno testato su 10 proteine di diversi livelli di complessità strutturale, ciascuna delle quali lega una grande varietà di piccole molecole (solitamente denominate ligandi). Hanno poi confrontato i loro risultati non solo con quelli di altri metodi corrispondenti, ma anche con i risultati di esperimenti di laboratorio, ed entrambi i confronti si sono rivelati molto favorevoli.
"Naturalmente non siamo gli unici a lavorare su questo aspetto. Esistono diversi metodi simili. Di solito, tuttavia, la loro velocità è compensata da una bassa precisione, mentre calcoli più accurati possono richiedere diversi giorni. I nostri metodi sono unici in quanto possono elaborare le informazioni su grandi sistemi molecolari in decine di minuti mantenendo i vantaggi di calcoli quanto-meccanici molto più impegnativi," spiega Jan Řezáč, autore corrispondente dell'articolo del gruppo Interazioni non covalenti guidato dal Prof. Pavel Hobza.
Gli esperti di questo gruppo studiano da molto tempo le interazioni intermolecolari. In questa ricerca si concentrano principalmente sulle biomolecole e i risultati del loro lavoro riguardano direttamente la progettazione assistita da computer dei farmaci. Il motivo è che quando gli scienziati lavorano su un nuovo farmaco, spesso cercano molecole che si legano fortemente a una particolare proteina.
Identificarle, tuttavia, è come trovare aghi in un pagliaio, poiché è necessario testare un gran numero di molecole per distinguere quelle che si dimostrano promettenti. Ciò rallenta notevolmente la scoperta delle sostanze medicinali e la rende più costosa. Predicendo la forza del legame proteina-ligando e quindi individuando le molecole che meglio soddisfano una serie di criteri definiti, i chimici computazionali risparmiano il lavoro degli sperimentatori, il che, a sua volta, accelera in modo significativo la scoperta di farmaci.
Ulteriori informazioni: Adam Pecina et al, SQM2.20:La funzione di punteggio quantomeccanico semiempirico produce previsioni di affinità di legame proteina-ligando di qualità DFT in pochi minuti, Nature Communications (2024). DOI:10.1038/s41467-024-45431-8
Informazioni sul giornale: Comunicazioni sulla natura
Fornito dall'Istituto di Chimica Organica e Biochimica del CAS