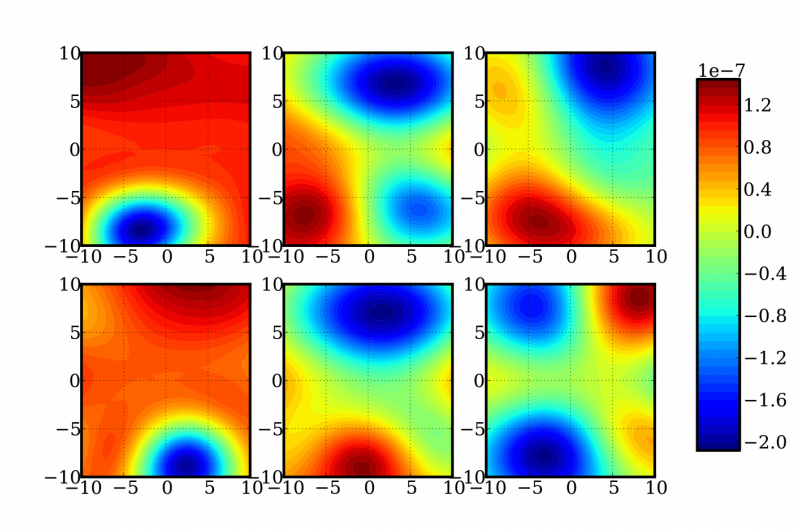
Una rappresentazione di fette bidimensionali casuali di una funzione a 12 dimensioni per determinare le correzioni di energia e frequenza di una molecola di formaldeide. Credito:Laboratori Nazionali Sandia
I ricercatori dei Sandia National Laboratories hanno sviluppato nuove tecniche matematiche per far progredire lo studio delle molecole a livello quantistico.
Sviluppi matematici e algoritmici in questa direzione sono necessari per consentire lo studio dettagliato di complesse molecole di idrocarburi che sono rilevanti nella combustione del motore.
I metodi esistenti per approssimare le potenziali funzioni energetiche su scala quantistica richiedono troppa potenza del computer e sono quindi limitati a piccole molecole. I ricercatori di Sandia affermano che la loro tecnica accelererà i calcoli della meccanica quantistica e migliorerà le previsioni fatte dai modelli di chimica teorica. Data la velocità di calcolo, questi metodi possono essere potenzialmente applicati a molecole più grandi.
Il ricercatore post-dottorato di Sandia Prashant Rai ha lavorato con i ricercatori Khachik Sargsyan e Habib Najm presso il Combustion Research Facility di Sandia e ha collaborato con i chimici quantistici So Hirata e Matthew Hermes presso l'Università dell'Illinois a Urbana-Champaign. Calcolo dell'energia con meno disposizioni geometriche di quelle normalmente richieste, il team ha sviluppato metodi computazionalmente efficienti per approssimare le potenziali superfici energetiche.
Una comprensione precisa delle potenziali superfici energetiche, elementi chiave in quasi tutti i calcoli della dinamica quantistica, è necessario per stimare con precisione l'energia e la frequenza dei modi vibrazionali delle molecole.
"Se riusciamo a trovare l'energia della molecola per tutte le possibili configurazioni, possiamo determinare informazioni importanti, come stati stabili della struttura di transizione molecolare o stati intermedi di molecole nelle reazioni chimiche, " ha detto Rai.
I primi risultati di questa ricerca sono stati pubblicati in Fisica Molecolare in un articolo intitolato "Decomposizione tensoriale canonico di basso rango di superfici di energia potenziale:applicazione alla teoria della funzione di Green vibrazionale diagrammatica basata su griglia".

Ricercatori dei Laboratori Nazionali Sandia Prashant Rai, sinistra, Habib Najm, centro, e Khachik Sargsyan discutono delle tecniche matematiche utilizzate per studiare il comportamento di grandi molecole su scala quantistica. Credito:Dino Vournas
"L'approssimazione delle potenziali superfici energetiche di molecole più grandi è un compito estremamente impegnativo a causa dell'aumento esponenziale delle informazioni necessarie per descriverle con ogni atomo aggiuntivo nel sistema, " ha detto la Rai. "In matematica, è chiamata la Maledizione della Dimensionalità."
Sconfiggere la maledizione
La chiave per vincere la maledizione della dimensionalità è sfruttare le caratteristiche della struttura specifica delle potenziali superfici energetiche. Rai ha affermato che queste informazioni sulla struttura possono quindi essere utilizzate per approssimare le funzioni ad alta dimensione richieste.
"Sfruttiamo il fatto che sebbene le superfici di energia potenziale possano essere ad alta dimensionalità, possono essere ben approssimati come una piccola somma di prodotti di funzioni unidimensionali. Questo è noto come la struttura di basso rango, dove il rango della superficie dell'energia potenziale è il numero di termini nella somma, «Un simile presupposto sulla struttura», ha detto la Rai, «è abbastanza generale ed è stato utilizzato anche in problemi simili in altri campi. Matematicamente, l'intuizione delle tecniche di approssimazione di basso rango deriva dall'algebra multilineare in cui la funzione viene interpretata come tensore e viene scomposta utilizzando tecniche di decomposizione tensoriale standard."
Le correzioni di energia e frequenza sono formulate come integrali di queste funzioni energetiche ad alta dimensionalità. L'approssimazione in un formato così basso rende queste funzioni facilmente integrabili in quanto rompe il problema dell'integrazione alla somma dei prodotti di integrali uno o due dimensioni, quindi si applicano metodi di integrazione standard.
Il team ha provato i propri metodi computazionali su piccole molecole come acqua e formaldeide. Rispetto al metodo classico Monte Carlo, il cavallo di battaglia standard basato sulla casualità per problemi di integrazione dimensionale elevati, il loro approccio prevedeva energia e frequenza della molecola d'acqua più accurate, ed era almeno 1, 000 volte più efficiente dal punto di vista computazionale.
Rai ha affermato che il prossimo passo è migliorare ulteriormente la tecnica sfidandola con molecole più grandi, come il benzene.
"Studi interdisciplinari, come la chimica quantistica e l'ingegneria della combustione, fornire opportunità per l'impollinazione incrociata di idee, fornendo così una nuova prospettiva sui problemi e sulle loro possibili soluzioni, "Ha detto Rai. "È anche un passo verso l'utilizzo dei recenti progressi nella scienza dei dati come pilastro della scoperta scientifica in futuro".