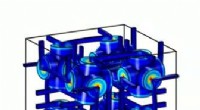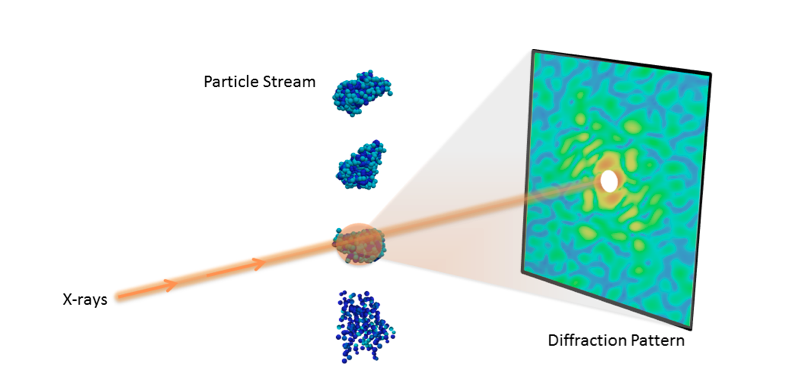
Configurazione sperimentale per un esperimento di diffrazione di una singola particella. Credito:Peter Zwart, Berkeley Lab
La comprensione della struttura molecolare 3D di importanti nanooggetti come proteine e virus è fondamentale in biologia e medicina. Con i recenti progressi nella tecnologia a raggi X, gli scienziati possono ora raccogliere immagini di diffrazione da singole particelle, consentendo infine ai ricercatori di visualizzare le molecole a temperatura ambiente.
Però, determinare la struttura 3D da questi esperimenti di diffrazione di singole particelle è un ostacolo significativo. Ad esempio, le attuali velocità di acquisizione dei dati sono molto limitanti, in genere risulta in meno di 10 utili istantanee al minuto, limitare la quantità di funzionalità che possono essere risolte. Inoltre, le immagini sono spesso altamente corrotte da rumore e altri artefatti sperimentali, rendendo difficile interpretare correttamente i dati.
Per affrontare queste sfide, un team di ricercatori del Lawrence Berkeley National Laboratory (Berkeley Lab) ha sviluppato un nuovo framework algoritmico chiamato multi-tiered iterative phasing (M-TIP) che utilizza tecniche matematiche avanzate per determinare la struttura molecolare 3D da insiemi molto sparsi di rumorosi, dati di una singola particella. Questo approccio consente essenzialmente ai ricercatori di estrarre più informazioni da esperimenti con dati limitati. Matematici applicati Jeffrey Donatelli e James Sethian, e il bioscienziato fisico Peter Zwart hanno introdotto questa struttura espandendo un algoritmo che avevano originariamente sviluppato per risolvere la ricostruzione da un esperimento di diffusione dei raggi X correlato, chiamato diffusione di raggi X di fluttuazione. Un documento che descrive il framework M-TIP è stato pubblicato il 26 giugno nel Atti dell'Accademia Nazionale delle Scienze .
"Questo approccio ha il potenziale per rivoluzionare il settore, " dice Zwart. "Dato che è difficile ottenere molti buoni dati, è probabile che gli approcci che riducono la quantità di dati necessari per visualizzare con successo i nanooggetti 3D ricevano un caloroso benvenuto".
Donatelli, Sethian e Zwart fanno tutti parte di CAMERA (The Center for Advanced Mathematics for Energy Research Applications), la cui missione è creare la matematica all'avanguardia necessaria per gestire i dati provenienti da molte delle strutture scientifiche più avanzate del DOE. CAMERA è finanziato congiuntamente dai programmi Advanced Scientific Computing Research e Basic Energy Sciences dell'Office of Science del DOE.
Diffrazione di singole particelle
Il recente avvento dei laser a elettroni liberi a raggi X (XFEL) ha consentito diverse nuove tecniche sperimentali per lo studio di biomolecole che non erano realizzabili con le sorgenti luminose tradizionali. Una di queste tecniche è la diffrazione di singole particelle, che raccoglie un gran numero di istantanee di diffrazione di raggi X con una sola particella nel raggio. Sfruttando l'estrema potenza degli XFEL, i ricercatori possono raccogliere segnali misurabili anche dalle particelle più piccole.
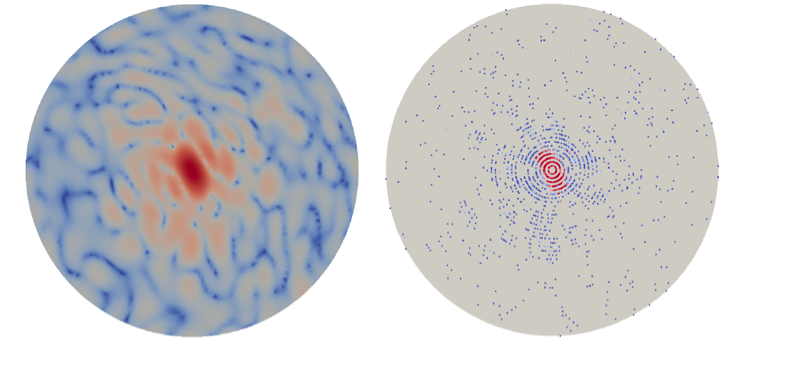
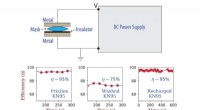
Un esempio di un'immagine di diffrazione di una singola particella pulita (a sinistra) e la stessa immagine di diffrazione dopo la contaminazione da rumore (a destra). Credito:Peter Zwart, Berkeley Lab
Un grande vantaggio offerto da questa tecnica di diffrazione di singole particelle è la capacità di studiare come le diverse copie di una molecola variano o cambiano forma. Poiché ogni immagine proviene da una singola particella, queste variazioni possono essere catturate nell'esperimento, in contrasto con i metodi di imaging tradizionali come la cristallografia o la diffusione di raggi X a piccolo angolo, dove i ricercatori possono misurare solo una media su tutti i diversi stati del campione molecolare.
Però, determinare la struttura 3D dai dati di diffrazione di singole particelle è impegnativo. Iniziare, quando ogni particella viene visualizzata, il suo orientamento è sconosciuto e deve essere recuperato per combinare correttamente i dati in un volume di diffrazione 3D. Questo problema è aggravato se la molecola può assumere forme diverse, che richiede una classificazione aggiuntiva delle immagini. Per di più, l'informazione di fase non viene registrata nelle immagini di diffrazione e deve essere recuperata per completare la ricostruzione. Finalmente, anche con potenti XFEL, il numero di fotoni sparsi è molto piccolo, con conseguente immagini estremamente rumorose, che può essere ulteriormente contaminato da problemi sistematici di fondo e di lettura del rivelatore.
Gli approcci precedenti si basano sulla risoluzione del problema della ricostruzione in passaggi separati, dove ogni singolo problema viene affrontato separatamente. Sfortunatamente, uno svantaggio di questi approcci seriali è che non sfruttano facilmente le caratteristiche note precedenti sull'aspetto della molecola. Inoltre, qualsiasi errore commesso in un passaggio viene propagato al successivo, determinando un ulteriore aumento dell'errore. Questo "errore a valanga" alla fine degrada la qualità della ricostruzione ottenuta nella fase finale.
Il meglio di entrambi i mondi
Invece di risolvere i problemi di calcolo in passaggi separati, l'algoritmo M-TIP del team risolve tutte le parti del problema contemporaneamente. Questo approccio sfrutta le informazioni precedenti sulla struttura per ridurre notevolmente i gradi di libertà del problema in tutte le fasi, e di conseguenza ridurre le informazioni necessarie per realizzare una ricostruzione 3D.
"Le tecniche standard di ottimizzazione della scatola nera possono incorporare le conoscenze pregresse nella ricostruzione ma gettare via tutta la struttura del problema, considerando che risolverlo in passaggi secondari seriali completamente separati sfrutta la struttura del problema ma elimina quasi tutte le informazioni precedenti su come potrebbe essere la soluzione, " ha detto Donatelli. "M-TIP sfrutta il meglio di entrambi i mondi sfruttando la struttura del problema per suddividere il calcolo in diversi blocchi gestibili e quindi perfezionando iterativamente tutti questi blocchi per arrivare a una soluzione che sia coerente con entrambi i dati ed eventuali vincoli strutturali."
Utilizzando questa tecnica, il team è stato in grado di determinare la struttura 3D da conteggi di immagini estremamente bassi da dati simulati, da 6 a 24 immagini per dati privi di rumore e 192 immagini da dati altamente contaminati.
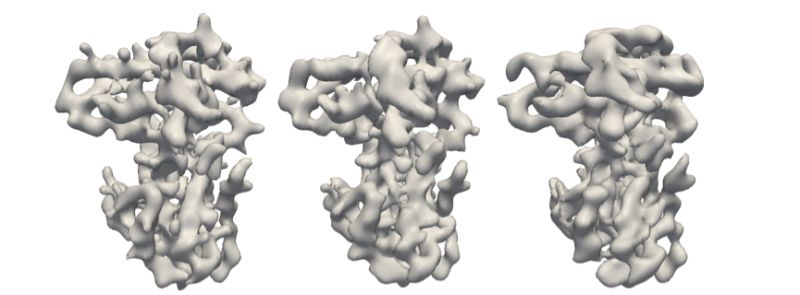
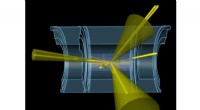
Proteina originale del retinoblastoma (a sinistra) e ricostruzioni utilizzando l'algoritmo M-TIP con 24 immagini pulite (al centro) e 192 immagini rumorose (a destra), come mostrato nella Figura 2. Credito:Peter Zwart, Berkeley Lab
Rompere un nuovo terreno
Questo lavoro fa parte di una nuova iniziativa di collaborazione tra SLAC National Accelerator Laboratory, TELECAMERA, il National Energy Research Scientific Computing Center (NERSC) e il Los Alamos National Laboratory come parte dell'Exascale Computing Project (ECP) del DOE. L'obiettivo del progetto è sviluppare gli strumenti computazionali necessari per eseguire l'analisi dei dati in tempo reale da esperimenti condotti presso la Linac Coherent Light Source (LCLS) di SLAC. Con gli aggiornamenti alla linea di luce, LCLS-II prevede di generare diversi terabyte di dati al secondo, quale, Per esempio, consentirà agli scienziati di ampliare notevolmente gli attuali esperimenti sulle singole particelle. L'analisi di tutti questi dati in tempo reale richiederà nuovi algoritmi e grandi macchine di calcolo. L'algoritmo M-TIP servirà come parte di questo processo.
"Questi sono alcuni dei problemi più impegnativi nella scienza dei dati computazionali, " dice Sethian. "Per affrontarli, dobbiamo sfruttare una gamma di tecnologie, comprese le emergenti architetture di elaborazione exascale, sofisticate reti ad alta velocità, e gli algoritmi matematici più avanzati disponibili. Riunire gli scienziati di CAMERA con progetti applicativi exascale ha aperto la porta alla creazione di strumenti per affrontare alcuni problemi urgenti in biologia e scienze dei materiali."
I ricercatori osservano che questi sono solo i primi passi. Affinché il metodo sia pronto per essere distribuito, altri ostacoli devono essere superati.
"La scienza sperimentale è disordinata, " dice Zwart. "Ci sono ulteriori effetti sperimentali che devono essere presi in considerazione per poter ottenere i migliori risultati possibili".
"Fortunatamente, M-TIP è una tecnica molto modulare, "aggiunge Donatelli, "così, è adatto a modellare molti di questi effetti aggiuntivi senza dover modificare il framework algoritmico di base".
Il team sta attualmente lavorando allo studio di questi effetti nell'ambito della Single Particle Initiative, un grande, collaborazione multi-istituzionale dedicata ad affrontare questioni teoriche e pratiche nell'imaging di singole molecole basato su X-FEL, portando infine a fornire alla comunità scientifica gli strumenti necessari per aprire nuovi orizzonti in biologia, medicina e scienze energetiche.