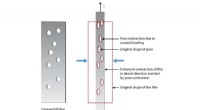
Modello strutturale della fibrilla peptidica amiloide A-beta 1-42 di Alzheimer derivata da una struttura sperimentale (PDB:2MXU). Gli aggregati fibrillari agiscono come tossine cellulari all'inizio e alla progressione della malattia di Alzheimer. Credito:Emanuel Peter
proteine, gli onnipresenti cavalli di battaglia della biochimica, sono enormi molecole la cui funzione dipende da come si ripiegano in strutture complesse. Per capire come funzionano queste molecole, i ricercatori usano la modellazione al computer per calcolare come si ripiegano le proteine.
Ora, un nuovo algoritmo può accelerare quelle simulazioni vitali, consentendo loro di modellare fenomeni che prima erano fuori portata. I risultati possono eventualmente aiutare gli scienziati a comprendere e curare meglio malattie come l'Alzheimer, disse Emanuele Pietro, un chimico presso l'Università di Ratisbona. Il suo lavoro sulla nuova tecnica è descritto questa settimana in Il Giornale di Fisica Chimica .
Simulazioni convenzionali, utilizzando la dinamica molecolare e metodi Monte Carlo, hanno avuto successo nel modellare molecole biologiche come le proteine. Per determinare come le proteine si ripiegano, la simulazione ricerca configurazioni che corrispondono a stati energetici sempre più bassi. Infine, trova lo stato energetico più basso, che dà una piega stabile. Ma mentre la simulazione cerca, può incontrare una configurazione con un'energia leggermente superiore, che forma una barriera che impedisce l'algoritmo.
A causa di questi rallentamenti, i metodi convenzionali possono solo simulare comportamenti molecolari che si verificano su scale temporali brevi di poche centinaia di microsecondi. Molti fenomeni, come alcune pieghe proteiche o un farmaco che si lega a un potenziale bersaglio, accadere nel giro di pochi secondi, minuti o addirittura giorni. La simulazione di tempi così lunghi richiederebbe troppo tempo di calcolo con solo approcci convenzionali.
Per velocizzare le simulazioni, i ricercatori possono iniettare energia nel sistema, che spinge il modello oltre ogni barriera energetica. Ma una delle maggiori sfide a questi metodi è nel definire le coordinate che descrivono il sistema, che, Per esempio, può essere la lunghezza tra gli atomi nella molecola, e gli angoli tra i legami. Tradizionalmente, i ricercatori definiscono le coordinate prima di iniziare la simulazione. Ogni passo lungo ogni coordinata dipende dal passo precedente. Ma questa dipendenza può influenzare la simulazione.
Il nuovo algoritmo di Peter evita questo pregiudizio. Ha trovato un sistema di coordinate generalizzato in cui ogni passaggio temporale non si basa sul passaggio precedente. "Sono necessari solo pochi parametri, e non è necessaria l'intuizione umana, che possono potenzialmente distorcere il risultato della simulazione, " Egli ha detto.
Per testare il nuovo algoritmo, Peter lo usava per modellare l'acqua, un peptide chiamato dialanina, il ripiegamento di un altro peptide chiamato TrpCage, e l'aggregazione di beta-amiloide 25-35, che sono frammenti proteici associati alla malattia di Alzheimer. In ogni caso, la sua tecnica riporta di aver velocizzato le simulazioni. E le simulazioni dell'amiloide-beta potrebbero aiutare a spiegare perché l'Alzheimer è stato difficile da curare.
Nella malattia di Alzheimer, frammenti di proteine beta-amiloide si aggregano insieme, formando una placca dura che si accumula tra i neuroni e li distrugge. L'amiloide-beta è anche una tossina, portando alla morte delle cellule neuronali e alla degenerazione della funzione neuronale. Le nuove simulazioni suggeriscono che l'amiloide-beta può assumere una serie di strutture. Questa flessibilità strutturale potrebbe essere il motivo per cui alcuni farmaci che cercano di inibire l'aggregazione non hanno avuto successo, ha detto Pietro. Quando quei farmaci si legano all'amiloide-beta, l'amiloide-beta cambia solo forma, permettendogli di continuare ad aggregarsi. Il farmaco viene incorporato nell'aggregato e nella placca.
Questo tipo di flessibilità strutturale, chiamata entropia di conformazione, è anche una caratteristica chiave in altri peptidi che formano placche tossiche in malattie come la malattia di Huntington, diabete di tipo 2, e il morbo di Parkinson. Il nuovo algoritmo potrebbe quindi essere utile per comprendere anche queste altre malattie.