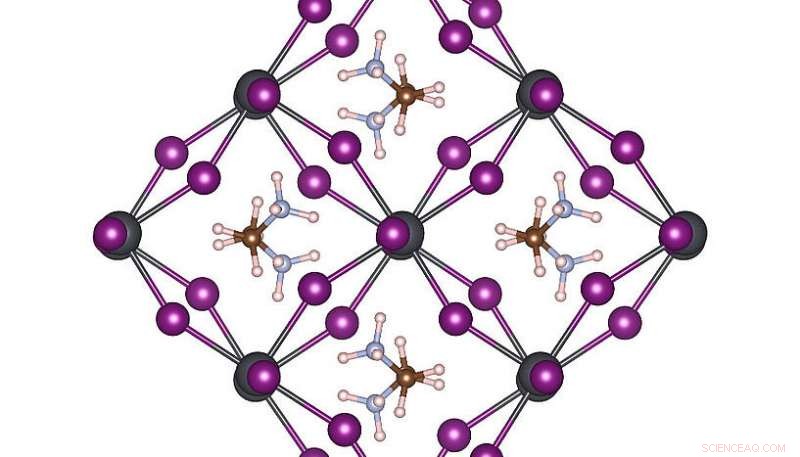
Struttura atomica ad alta simmetria di MAPbI3 a temperatura ambiente. Credito:Menno Bokdam/Università di Vienna
I materiali su scala atomica possono mostrare una ricca gamma di comportamenti dinamici, che influenza direttamente le proprietà fisiche di questi materiali. Per molti anni, è stato un sogno descrivere queste dinamiche in materiali complessi a varie temperature utilizzando simulazioni al computer. I fisici dell'Università di Vienna hanno sviluppato un metodo di apprendimento automatico al volo che consente tali calcoli attraverso l'integrazione diretta nel Vienna Ab-initio Simulation Package (VASP) basato sulla meccanica quantistica. La versatilità del metodo di autoapprendimento è dimostrata da nuove scoperte, pubblicato sulla rivista Lettere di revisione fisica , sulle transizioni di fase delle perovskiti ibride. Queste perovskiti sono di grande interesse scientifico per il loro potenziale nella raccolta dell'energia solare e in altre applicazioni.
A temperatura ambiente, tutti i materiali sono in costante movimento su scala atomica. Anche la roccia solida è costituita da atomi che oscillano. Le proprietà fisiche dei materiali sono direttamente legate alla disposizione degli atomi nella, così chiamato, reticolo cristallino. A seconda della temperatura o della pressione questa disposizione può cambiare influenzando così le proprietà dei materiali. Si può pensare al diamante, che è trasparente e duro a causa della disposizione periodica degli atomi di carbonio nel cristallo di diamante. Gli stessi atomi, disposti in modo diverso, risulta nero, grafite fragile. Era già possibile calcolare con precisione le coordinate degli atomi in materiali semplici a diverse temperature con simulazioni di dinamica molecolare quantistica (MD). Però, tali calcoli sono computazionalmente costosi e limitano le applicazioni pratiche a un paio di centinaia di atomi e un tempo di simulazione limitato.
I fisici del gruppo Computational Materials Physics dell'Università di Vienna hanno sviluppato un nuovo approccio che supera queste limitazioni e rende possibili simulazioni di materiali complessi per future applicazioni energetiche. Ciò si ottiene sviluppando un algoritmo di autoapprendimento basato sui dati efficiente e robusto e, più importante, integrando questo algoritmo direttamente nel Vienna Ab-initio Simulation Package (VASP). Nel nuovo approccio, la "macchina" può prelevare, da solo, gli ingredienti essenziali per una descrizione del modello più semplice degli atomi interagenti durante le simulazioni MD. Già dopo aver calcolato alcune centinaia di passi temporali la macchina può prevedere con sufficiente precisione le posizioni degli atomi nel passo temporale consecutivo. La macchina è anche in grado di effettuare una stima della sua precisione per i passaggi successivi. Se l'errore è troppo alto, la macchina cambia marcia ed esegue la precisione, ma costoso, calcoli MD. Più passa il tempo di simulazione, più la macchina apprende e più precisa diventa. In questo modo, sono richiesti sempre meno calcoli MD, che alla fine porta alla situazione in cui tutti i passaggi temporali sono fatti dalla macchina. Inoltre, la capacità di autoapprendimento al volo riduce la necessità di intervento umano richiesto da altri metodi di apprendimento automatico esistenti.
Per dimostrare la potenza di questo nuovo metodo, i ricercatori lo hanno applicato per studiare le transizioni tra le diverse strutture atomiche del MAPbI 3 perovskite al variare della temperatura. Questo materiale è molto popolare per il suo potenziale come nuovo componente di celle solari a basso costo. È fatto di molecole organiche che possono capovolgersi rapidamente, separati l'uno dall'altro da un reticolo composto da atomi di piombo e ioduro. A seconda della temperatura si formano tre diverse fasi cristalline. I meccanismi atomici vicino alla temperatura di transizione sono molto difficili da determinare sperimentalmente, e le simulazioni MD richiederebbero anni di tempo di calcolo anche su un moderno sistema di supercalcolo. Dopo aver appreso, la macchina può prevedere le temperature di transizione di fase e le costanti reticolari di questo materiale con una precisione senza precedenti. Il metodo sviluppato è generale e applicabile a molti altri futuri problemi di scienza dei materiali e sarà disponibile per i ricercatori di tutto il mondo nella prossima versione di VASP.
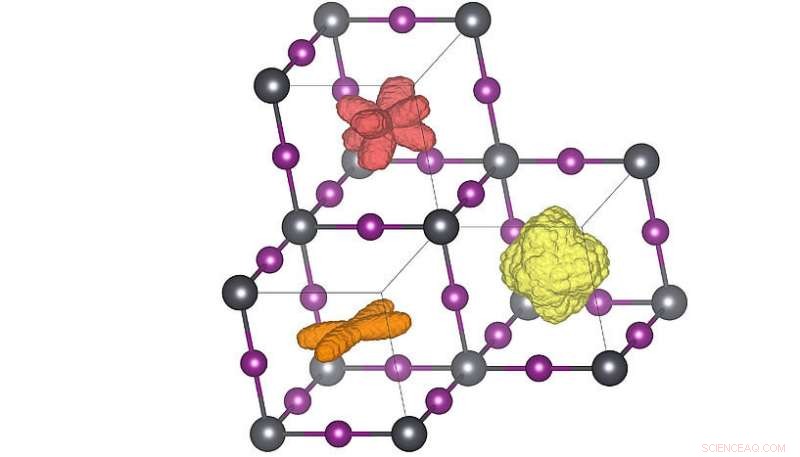
Distribuzioni tridimensionali dell'orientamento della molecola nelle tre diverse fasi cristalline. Quando la temperatura è elevata (arancione→ rosso→ giallo) le molecole possono raggiungere più orientamenti. La distribuzione rossa corrisponde alla struttura della temperatura ambiente. Credito:Menno Bokdam/Università di Vienna