Credito:Weng Hong Sio.
Un team di ricercatori dell'Università di Oxford ha recentemente introdotto un nuovo modo per modellare i polaroni, una quasiparticella tipicamente utilizzata dai fisici per comprendere le interazioni tra elettroni e atomi nei materiali solidi. Il loro metodo, presentato in un articolo pubblicato in Lettere di revisione fisica , combina modelli teorici con simulazioni computazionali, consentendo osservazioni approfondite di queste quasiparticelle in un'ampia gamma di materiali.
Essenzialmente, un polarone è una particella composita composta da un elettrone circondato da una nuvola di fononi (cioè vibrazioni reticolari). Questa quasiparticella è più pesante dell'elettrone stesso ea causa del suo peso sostanziale può talvolta rimanere intrappolata in un reticolo cristallino.
I polaroni contribuiscono alla corrente elettrica che alimenta diversi strumenti tecnologici, compresi diodi organici a emissione di luce e touchscreen. Comprendere le loro proprietà è quindi di fondamentale importanza, in quanto potrebbe aiutare a sviluppare la prossima generazione di vari dispositivi per l'illuminazione e l'optoelettronica.
"Il lavoro precedente sui polaroni si basava su modelli matematici idealizzati, "Prof. Feliciano Giustino, il capo del gruppo che ha svolto lo studio, ha detto a Phys.org. "Questi modelli sono stati molto utili per comprendere le proprietà di base dei polaroni, ma non tengono conto della struttura dei materiali su scala atomica, quindi non sono sufficienti quando si cerca di studiare materiali reali per applicazioni pratiche. La nostra idea era di sviluppare una metodologia computazionale che consentisse indagini sistematiche sui polaroni con accuratezza predittiva".
Il metodo ideato dal team di Giustino si basa sulla teoria del funzionale densità, che è attualmente lo strumento più popolare per la modellazione e la progettazione predittiva dei materiali utilizzando la meccanica quantistica. Una delle principali sfide incontrate quando si studiano i polaroni basati su questa teoria è che le risorse computazionali richieste (ore CPU) sono proporzionali alla terza potenza del numero di atomi da simulare. In altre parole, se si studiassero due cristalli con 10 e 20 atomi per cella unitaria, il calcolo richiesto per studiare il secondo cristallo richiederebbe 8 volte più tempo di quello richiesto per il primo.
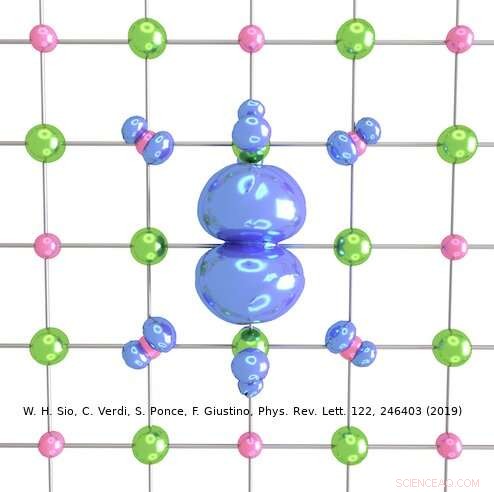
Credito:Weng Hong Sio.
Poiché molti polaroni hanno una dimensione di 1-2 nanometri, i calcoli per studiare questi sistemi richiederebbero celle di simulazione con almeno 3, 000-5, 000 atomi. Eppure le attuali capacità di calcolo farebbero fatica a sostenere tali simulazioni e ciascuno dei numerosi calcoli necessari per indagare su questi sistemi richiederebbe settimane, anche quando si utilizza un moderno supercomputer.
"La nostra idea era di cercare di rendere questo processo più efficiente sfruttando i progressi nella cosiddetta teoria delle perturbazioni densità-funzionale, " Weng Hong Sio, il primo autore dell'opera, spiegato. "Senza entrare nei dettagli, siamo stati in grado di riformulare il problema di eseguire un calcolo di un polarone in una grande cella di simulazione nel problema più semplice di eseguire calcoli multipli nella cella unitaria più piccola del cristallo. Questa strategia ha aperto nuove possibilità che prima erano inaccessibili".
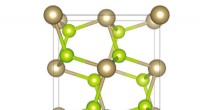
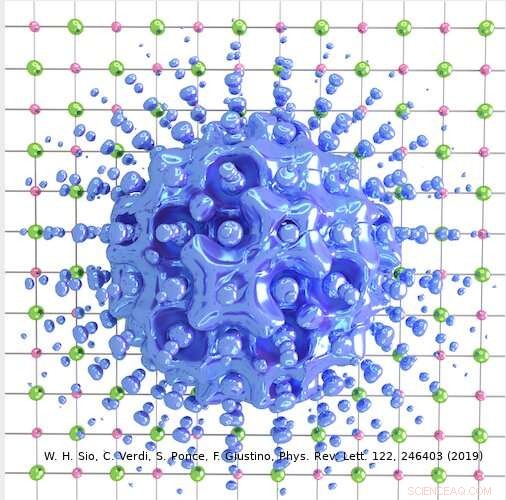
L'approccio ideato dal team di Giustino può essere utilizzato per descrivere sia polaroni grandi che piccoli. Nel loro studio, ad esempio, i ricercatori hanno mostrato come può essere utilizzato per calcolare le funzioni d'onda, energie di formazione e decomposizione spettrale dei polaroni in LiF e Li 2 oh 2 composti. Utilizzando il loro metodo di simulazione, hanno scoperto che i polaroni nei sali semplici e negli ossidi metallici utilizzati nelle batterie hanno una struttura interna molto più ricca di quanto suggerito da precedenti lavori sul campo.
"Per esempio, nel sale prototipo fluoruro di litio, in precedenza si pensava che il polarone nascesse dall'interazione tra un elettrone e fononi ottici longitudinali, cioè le vibrazioni reticolari responsabili della risposta dielettrica del cristallo, " Spiegò Sio. "Abbiamo scoperto che questi non sono gli unici fononi coinvolti, e che anche l'interazione tra l'elettrone e i fononi piezoacustici (cioè le vibrazioni responsabili della piezoelettricità) è importante."
Le osservazioni raccolte dal team di Giustino cambiano l'attuale prospettiva sui polaroni nel sale di litio fouride, che è un sistema molto semplice. Applicare il loro metodo a sistemi più complessi potrebbe svelare strutture ancora più ricche, migliorando infine la nostra attuale comprensione delle loro proprietà e informando lo sviluppo di nuovi materiali con proprietà polatroniche su misura. Nelle loro ricerche future, i ricercatori intendono utilizzare il loro metodo per studiare altri materiali, al fine di valutare ulteriormente il suo potere predittivo e ottenere una migliore comprensione di altri materiali tecnologicamente importanti.
"Più avanti sarà importante indagare su cosa può fare un polarone:per ora sappiamo che possiamo calcolare la configurazione a più bassa energia di un polarone, ma non abbiamo idea di cosa succede se questo polarone è soggetto a campi elettrici o magnetici statici o a radiazioni elettromagnetiche, — disse Giustino. — Inoltre, strette interazioni con i gruppi sperimentali saranno essenziali per tradurre questi risultati in applicazioni".
© 2019 Scienza X Rete