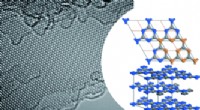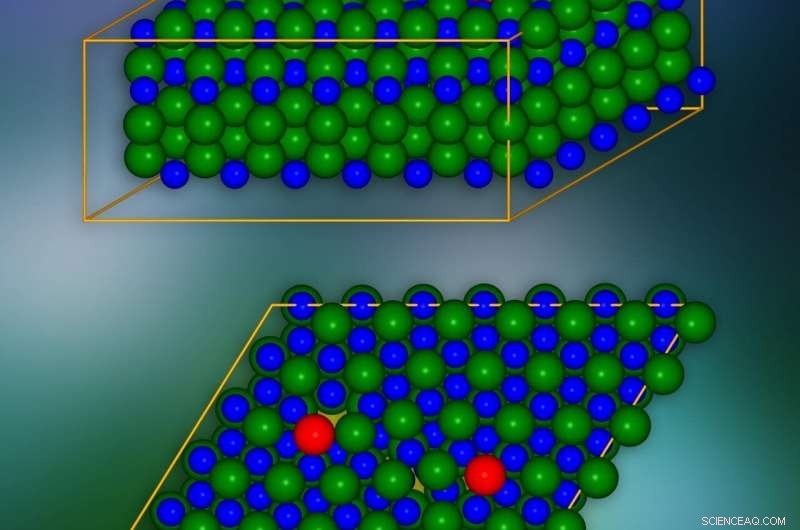
Modello in cristallo al carburo di silicio con dislocazioni di spigolo introdotte nei punti segnati in rosso. In basso è presentato un unico piano cristallografico. I punti in cui le cariche elettriche possono "fuoriuscita" dagli strati vicini sono contrassegnati in giallo. Credito:IFJ PAN
Imperfezioni della struttura cristallina, in particolare dislocazioni di bordo di natura allungata, modificare profondamente le proprietà di base dell'intero materiale e, conseguentemente, limitare drasticamente le sue applicazioni. Prendendo come esempio il carburo di silicio, fisici di Cracovia e Varsavia hanno dimostrato che anche tali difetti computazionalmente impegnativi possono essere esaminati con successo con accuratezza atomica per mezzo di un abilmente costruito, di piccole dimensioni, modello.
La matematica ama la perfezione. Sfortunatamente, la perfezione non ama la realtà fisica. I teorici che modellano i cristalli hanno a lungo cercato di includere i difetti nelle strutture cristalline reali e di prevederne l'impatto sulle proprietà fisiche dei materiali. I modelli, sulla base dei risultati di vari esperimenti, hanno descritto cambiamenti nelle proprietà di base di un materiale senza spiegare le reali cause ed effetti dei fenomeni che si verificano.
Un nuovo modello di carburo di silicio (SiC), costruito da fisici dell'Istituto di fisica nucleare dell'Accademia polacca delle scienze (IFJ PAN) a Cracovia, ha permesso loro di dimostrare che è ora possibile studiare i cristalli ab initio con difetti così complessi come le dislocazioni dei bordi e di spiegare le loro caratteristiche con processi che avvengono su scala atomica. Questo risultato spettacolare, recentemente presentato alla conferenza Multiscale Phenomena in Molecular Matter 2019 a Cracovia, è stato realizzato dai fisici IFJ PAN in collaborazione con l'Istituto di ricerca tecnologica fondamentale dell'Accademia polacca delle scienze e l'Istituto di fisica ad alta pressione dell'Accademia polacca delle scienze, entrambi situati a Varsavia.
"Abbiamo cercato di trovare i meccanismi responsabili a livello atomico per abbassare la tensione di rottura nei cristalli di carburo di silicio. I nostri calcoli ab initio portano a una comprensione qualitativa del problema e contribuiscono a spiegare i dettagli di questo fenomeno, "dice il dottor Jan Lazewski, professore all'IFJ PAN.
I calcoli ab initio hanno ormai una lunga storia legata al premio Nobel per Walter Kohn e John Pople nel 1998 (tuttavia, le simulazioni di difetti lineari dei cristalli sono state introdotte solo di recente). Questo termine è usato per descrivere calcoli eseguiti utilizzando equazioni della meccanica quantistica, supportato solo dalla conoscenza della struttura dell'atomo e della simmetria dei cristalli. Non ci sono informazioni dirette da esperimenti in tali modelli, il che significa che possono essere utilizzati anche per analizzare materiali che non sono mai stati studiati o addirittura sintetizzati prima. A causa della complicazione relativamente sostanziale del problema, finora i calcoli ab initio hanno funzionato, al massimo, in caso di difetti puntuali, relativi a vacanze (atomi mancanti o buchi nella struttura cristallina) e miscele introdotte nel cristallo.
Non senza motivo i ricercatori di Cracovia hanno utilizzato il carburo di silicio. Le proprietà di questo semiconduttore sono così interessanti che in passato era addirittura considerato un successore del silicio. Il suo band gap (la barriera che la carica deve superare per passare dalla banda di valenza alla banda di conduzione e condurre la corrente) è quasi tre volte maggiore che nel silicio, la densità di corrente di conduzione ammissibile:il doppio, la capacità di dissipare il calore, più di tre volte maggiore, e la frequenza di taglio del funzionamento del cristallo fino a sei volte maggiore. Inoltre, i sistemi al carburo di silicio possono funzionare a temperature fino a 650 gradi Celsius, mentre i sistemi al silicio iniziano già ad avere problemi a 120 gradi Celsius. SiC ha anche un alto punto di fusione, è difficile, resistente agli acidi e alle radiazioni. Tra i suoi svantaggi c'è soprattutto il prezzo:mentre i wafer di silicio da due pollici costano solo pochi dollari, il valore di wafer di carburo di silicio simili è di migliaia. I cristalli di carburo di silicio di bassa qualità sono un materiale abrasivo popolare, utilizzato anche nei giubbotti antiproiettile e nei dischi freno delle auto più costose del mondo, come Lamborghini o Bugatti. Cristalli di alta qualità vengono utilizzati per produrre specchi per telescopi e in dispositivi ad alta tensione con elevata resistenza alla temperatura.
A livello atomico, i cristalli di carburo di silicio sono composti da molti strati piatti disposti uno sopra l'altro. Ogni strato ricorda un nido d'ape:è costituito da celle esagonali in cui le molecole di carburo di silicio sono disposte verticalmente negli angoli. Ciascuno dei due strati adiacenti può essere combinato in tre modi. I 'sandwich' multistrato con diversi layout creano i cosiddetti politipi, di cui ne esistono più di 250 nel caso del carburo di silicio. Il gruppo di IFJ PAN ha utilizzato il polimorfo 4H-SiC.
"Quando si modellano tali strutture, uno dei problemi principali è la complessità computazionale. Un modello di puro cristallo, privo di mescolanze o dislocazioni, è caratterizzato da un'elevata simmetria e può essere calcolato anche in pochi minuti. Per eseguire un calcolo per un materiale con dislocazione, abbiamo bisogno di mesi di lavoro su un computer ad alta potenza, " sottolinea il dottor Pawel Jochym, professore all'IFJ PAN.
I problemi con le dislocazioni dei bordi derivano dalla scala della loro influenza sulla struttura cristallina del materiale. Come un'illustrazione, possono essere paragonati al problema di mascherare uno spazio vuoto in una fila di piastrelle su un pavimento. Il divario può essere "mimetizzato" spostando le tessere delle file adiacenti, ma il difetto rimarrà sempre visibile. Le dislocazioni dei bordi derivanti dalla mancanza di intere lunghezze o regioni di atomi/molecole nei singoli strati di cristallo agiscono in modo simile, influenzare le posizioni di atomi e molecole in molti strati adiacenti. E poiché le dislocazioni possono estendersi su lunghe distanze, in pratica i disturbi da essi provocati comprendono l'intero cristallo.
I fenomeni più interessanti si verificano nel nucleo di dislocazione, cioè in prossimità del bordo dello strato danneggiato della rete cristallina. Al fine di eliminare gli effetti a lungo raggio causati da una singola dislocazione, e quindi ridurre significativamente il numero di atomi in esame, è stato impiegato un trucco:è stata introdotta una seconda dislocazione dell'effetto opposto. In questo modo, l'impatto della prima dislocazione su lunghe distanze è stato compensato.
Il modello di cristallo di SiC consisteva di circa 400 atomi. Le simulazioni hanno mostrato che negli strati di cristalli, lungo il bordo del nucleo del difetto, i 'tunnel' appaiono sotto forma di canali con ridotta densità di carica. Abbassano la barriera potenziale localmente e provocano la "fuga" di cariche elettriche dalla banda di valenza. Inoltre, nel vuoto proibito, che nell'isolante garantisce una mancanza di conducibilità elettrica, compaiono condizioni che ne riducono l'ampiezza e l'efficacia nel limitare il flusso di carica. È stato dimostrato che questi stati provengono da atomi situati nel nucleo di dislocazione.
"La situazione può essere paragonata a un profondo, ripido burrone che uno scoiattolo sta cercando di attraversare. Se il fondo del burrone è vuoto, lo scoiattolo non arriverà dall'altra parte. Però, se c'è un numero di alberi in fondo abbastanza alto, lo scoiattolo può saltare sopra le loro cime dall'altra parte del burrone. Nel cristallo che abbiamo modellato, gli scoiattoli sono le cariche elettriche, la fascia di valenza è un bordo del burrone, la banda di conduzione è l'altra, e gli alberi sono i suddetti stati associati agli atomi del nucleo di dislocazione, " dice il prof. Lazewski.
Ora che i meccanismi responsabili dell'abbassamento della soglia della barriera energetica sono diventati noti a livello atomico, c'è un enorme margine di sperimentazione. Il meccanismo proposto dovrà essere verificato per poterlo utilizzare per limitare l'influenza negativa dei difetti testati. Fortunatamente, ci sono già possibilità tecniche per questo.
"Il futuro verificherà se le nostre idee saranno confermate nella loro interezza. Tuttavia, siamo fiduciosi sul destino del nostro modello e sull'approccio presentato alla simulazione delle dislocazioni dei bordi. Sappiamo già che il modello ab initio ha dimostrato la sua validità nel confronto con alcuni dati sperimentali, " conclude il prof. Jochym.