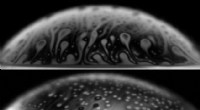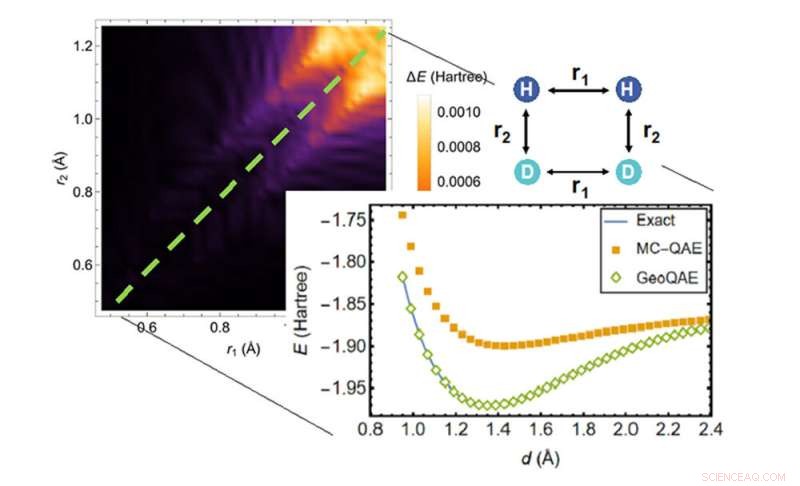
Nel calcolare la superficie dell'energia potenziale della reazione chimica di H2;+ D2 → 2HD, il nuovo algoritmo (diamanti verdi) supera l'algoritmo precedente (quadrati arancioni) nel trovare la soluzione più accurata (linea blu). Credito:Brookhaven National Laboratory
Un team di ricercatori del Brookhaven National Laboratory del Dipartimento dell'Energia degli Stati Uniti (DOE) e della Stony Brook University hanno ideato un nuovo algoritmo quantistico per calcolare le energie più basse delle molecole in configurazioni specifiche durante le reazioni chimiche, anche quando i loro legami chimici vengono rotti. Come descritto in Ricerca di revisione fisica , rispetto ad algoritmi esistenti simili, compreso il metodo precedente del team, il nuovo algoritmo migliorerà significativamente la capacità degli scienziati di calcolare in modo accurato e affidabile la superficie di energia potenziale nelle molecole che reagiscono.
Per questo lavoro, Deyu Lu, fisico del Center for Functional Nanomaterials (CFN) presso il Brookhaven Lab, ha lavorato con Tzu-Chieh Wei, professore associato specializzato in scienze dell'informazione quantistica presso il C.N. Yang Institute for Theoretical Physics presso la Stony Brook University, Qin Wu, un teorico al CFN, e Hongye Yu, un dottorato di ricerca. studente presso Stony Brook.
"Capire la meccanica quantistica di una molecola, come si comporta a livello atomico, può fornire informazioni chiave sulle sue proprietà chimiche, come la sua stabilità e reattività", ha affermato Lu.
Una proprietà particolare che è stata una sfida da determinare è lo stato fondamentale di una molecola:il punto in cui l'energia elettronica totale della molecola (inclusa l'energia cinetica e potenziale) è al suo livello più basso e nulla al di fuori di quel "sistema molecolare" è eccitante o carica elettroni. Quando la struttura atomica di un sistema chimico diventa più complessa, come in una grande molecola, molti più elettroni possono interagire. Tali interazioni rendono estremamente difficile il calcolo dello stato fondamentale di molecole complesse.
Il nuovo algoritmo quantistico migliora l'algoritmo precedente per affrontare questo problema in modo creativo. Sfrutta una deformazione geometrica regolare ottenuta variando continuamente le lunghezze di legame o gli angoli di legame nella struttura della molecola. Con questo approccio gli scienziati affermano di poter calcolare lo stato fondamentale delle molecole in modo molto accurato, anche se i legami chimici si rompono e si riformano durante le reazioni chimiche.
Costruire le basi
"Quando ci si basa esclusivamente sui metodi di calcolo tradizionali, questo problema dello stato fondamentale contiene troppe variabili da risolvere, anche sui supercomputer più potenti", ha affermato Lu.
Puoi pensare a un algoritmo come a un insieme di passaggi per risolvere un particolare problema. I computer classici possono eseguire algoritmi complessi, ma man mano che diventano più grandi e coinvolti, possono diventare troppo difficili o richiedere molto tempo per essere risolti in modo fattibile dai computer classici. I computer quantistici possono accelerare il processo sfruttando le regole della meccanica quantistica.
Nell'informatica classica, i dati sono archiviati in bit che hanno un valore di 1 o 0. Un bit quantistico, noto come qubit, può avere un valore superiore a 0 o 1, può anche avere un valore di 0 e 1, in un cosiddetta sovrapposizione quantistica. In linea di principio, questi qubit più "flessibili" possono memorizzare una quantità maggiore di informazioni rispetto ai bit classici. Se gli scienziati riescono a trovare il modo di sfruttare la capacità di trasporto di informazioni dei qubit, la potenza di calcolo può espandersi in modo esponenziale con ogni qubit aggiuntivo.
I qubit, tuttavia, sono piuttosto fragili. Spesso possono rompersi quando le informazioni vengono estratte. Quando un dispositivo quantistico interagisce con l'ambiente circostante, può generare rumore o interferenze che distruggono lo stato quantistico. Anche le variazioni di temperatura, le vibrazioni, le interferenze elettromagnetiche e persino i difetti dei materiali possono causare la perdita di informazioni dei qubit.
Per compensare queste insidie, gli scienziati hanno sviluppato una soluzione ibrida che sfrutta entrambi gli algoritmi di calcolo classici, che sono più stabili e pratici.
Lu e Wei hanno iniziato a ricercare sugli approcci ibridi di calcolo classico e quantistico nel 2019. Questa sovvenzione annuale promuove la collaborazione tra il Brookhaven National Laboratory e la Stony Brook University finanziando iniziative di ricerca congiunte in linea con le missioni di entrambe le istituzioni. Con questo lavoro iniziale, Lu e Wei si sono concentrati per la prima volta sulla risoluzione del problema dello stato fondamentale sostituendo gli algoritmi classici più "costosi" - quelli che erano molto più complessi e richiedevano significativamente più passaggi (e più tempo di calcolo) per essere completati - con quelli quantistici .
Allungare i legami, creare nuovi percorsi
I ricercatori osservano che gli algoritmi quantistici esistenti presentano tutti degli svantaggi per risolvere il problema dello stato fondamentale, incluso quello sviluppato da Wei e Yu nel 2019. Mentre alcuni algoritmi popolari sono accurati quando una molecola si trova nella sua geometria di equilibrio, la sua disposizione naturale degli atomi in tre dimensioni:questi algoritmi possono diventare inaffidabili quando i legami chimici vengono rotti a grandi distanze atomiche. La formazione e la dissociazione del legame svolgono un ruolo in molte applicazioni, come la previsione di quanta energia è necessaria per avviare una reazione chimica, quindi gli scienziati avevano bisogno di un modo per affrontare questo problema mentre le molecole reagiscono. Avevano bisogno di nuovi algoritmi quantistici in grado di descrivere la rottura del legame.
Per questa nuova versione dell'algoritmo, il team ha collaborato con il Brookhaven-Lab Co-design Center for Quantum Advantage (C2QA), formato nel 2020. Wei contribuisce alla spinta software del centro, specializzato in algoritmi quantistici. Il nuovo algoritmo del team utilizza un approccio adiabatico, che apporta cambiamenti graduali, ma con alcuni adattamenti che garantiscono che rimanga affidabile anche quando i legami chimici vengono rotti.
"Un processo adiabatico funziona adattando gradualmente le condizioni di un sistema quantomeccanico", ha spiegato Lu. "In un certo senso, stai raggiungendo una soluzione in passi molto piccoli. Fai evolvere il sistema da un modello semplice e risolvibile all'obiettivo finale, in genere un modello più difficile. Oltre allo stato fondamentale, tuttavia, un sistema multi-elettronico ha molti stati eccitati a energie più elevate. Questi stati eccitati possono rappresentare una sfida quando si utilizza questo metodo per calcolare lo stato fondamentale."
Wei ha paragonato un algoritmo adiabatico alla guida lungo un'autostrada, "se viaggi da una città all'altra, ci sono diversi percorsi per arrivarci, ma vuoi trovare quello più sicuro ed efficiente."
Nel caso della chimica quantistica, la chiave è trovare un "divario energetico" sufficientemente grande tra lo stato fondamentale e gli stati eccitati in cui non esistono stati di elettroni. Con uno spazio sufficientemente ampio, i veicoli nella metafora dell'autostrada non "attraverseranno corsie", quindi i loro percorsi possono essere tracciati con precisione.
"Un grande divario significa che puoi andare più veloce, quindi, in un certo senso, stai cercando di trovare un'autostrada meno affollata per guidare più velocemente senza colpire nulla", ha detto Wei.
"Con questi algoritmi, l'ingresso del percorso è una soluzione semplice e ben definita dell'informatica classica", ha osservato Wei. "Sappiamo anche dove si trova l'uscita, lo stato fondamentale della molecola, e stavamo cercando di trovare un modo per collegarla all'ingresso nel modo più naturale, una linea retta.
"L'abbiamo fatto nel nostro primo articolo, ma la linea retta presentava dei blocchi stradali causati dalla chiusura del gap energetico e dall'incrocio dei percorsi. Ora abbiamo una soluzione migliore."
Quando gli scienziati hanno testato l'algoritmo, hanno dimostrato che anche con variazioni finite della lunghezza del legame, la versione migliorata si comportava comunque in modo accurato per lo stato fondamentale.
"Siamo andati oltre la nostra zona di comfort, perché la chimica non è il nostro obiettivo", ha detto Wei. "Ma è stato bello trovare un'applicazione come questa e promuovere questo tipo di collaborazione con CFN. È importante avere prospettive diverse nella ricerca."
Ha notato lo sforzo accumulato di molte persone. "Nel grande schema, penso che stiamo dando un piccolo contributo, ma questo potrebbe essere una base per altri lavori in questi campi", ha detto. "Questa ricerca non è solo fondamentale, ma è un'ottima illustrazione di come diverse istituzioni e strutture possono unirsi per sfruttare le loro aree di competenza". + Esplora ulteriormente