La responsabile del progetto CNST Rachel Cannara e i collaboratori dell'Accademia Navale degli Stati Uniti (USNA) e dell'Università della Pennsylvania hanno dimostrato che la rugosità superficiale su scala atomica ha una forte influenza sull'adesione del diamante, carbonio amorfo, e modellare i nanocompositi di diamante.
Utilizzando misurazioni al microscopio a forza atomica (AFM) eseguite presso l'Università del Wisconsin-Madison, simulazioni di dinamica molecolare (MD), e teoria del funzionale della densità ab initio (DFT), hanno studiato la fisica e la meccanica dell'adesivo delle interfacce su scala nanometrica tra superfici di diamante.
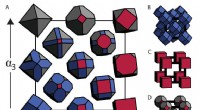
Per superfici atomicamente lisce, ci si aspetterebbe che la maggiore densità di atomi nel piano (111) porti a un momento di dipolo elettrostatico più elevato per unità di area ea un lavoro di adesione maggiore rispetto all'orientamento (001). Però, le misurazioni AFM, supportato da simulazioni dettagliate di nanocompositi modello di diamante, sfidare questa ipotesi in un modo che può essere spiegato solo dalle variazioni della rugosità superficiale a livello atomico, che per i cristalli singoli può derivare da meccanismi di crescita dipendenti dall'orientamento.
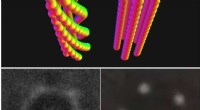
A differenza dei precedenti studi ab initio che hanno confrontato le energie superficiali per le superfici del diamante (111)(1×1)-H e le superfici del diamante non ricostruito (001)(1×1)-H, le simulazioni MD eseguite all'USNA simulano la superficie C(001)-H ricostruita (2×1). Le simulazioni prevedono che la superficie C(001)(2x1)-H sia energeticamente favorevole alla superficie non ricostruita. A conferma di queste simulazioni, le immagini della forza laterale AFM ad alta precisione della superficie (001) hanno rivelato (2 × 1) domini di riga di dimero.
Oltre a utilizzare la struttura della superficie appropriata (001), le simulazioni MD tengono conto delle interazioni di van der Waals a lungo raggio, così come le energie di superficie, nel calcolo del lavoro di adesione per ciascuna interfaccia. Inoltre, i calcoli ab initio DFT rivelano la presenza di dipoli di legame su superfici di diamante a cristallo singolo.
Utilizzando AFM, la meccanica di contatto dell'interfaccia è stata estratta dalla dipendenza dal carico dell'area di contatto durante gli esperimenti di attrito radente. I lavori di adesione sono stati quindi calcolati dal modello di meccanica di contatto appropriato e dalle forze di trazione misurate durante gli esperimenti di scorrimento e le misurazioni di forza-spostamento quasi statiche. Questi risultati hanno ampie implicazioni per la progettazione di dispositivi MEMS/NEMS che incorporano diamanti o materiali simili al diamante.