Figura 1:Angolo diedro (l'angolo formato dal piano creato dagli atomi A, B, e C, e il piano creato dagli atomi B, C, e D). Credito:Fujitsu
Fujitsu Laboratories ha annunciato oggi lo sviluppo di una tecnologia di simulazione molecolare per la scoperta di farmaci in grado di stimare con precisione l'affinità di legame, che rappresenta il grado in cui le proteine che possono causare malattie (proteine bersaglio) si legano a sostanze chimiche che potrebbero diventare farmaci candidati. Nel processo di scoperta di farmaci, è richiesta una previsione accurata dell'affinità di legame tra proteine bersaglio e sostanze chimiche, che offre una stima approssimativa dell'efficacia di un farmaco. La tecnologia di simulazione molecolare è stata ampiamente utilizzata in passato come metodo per prevedere l'affinità di legame, calcolare le forze approssimative che sorgono tra gli atomi nelle molecole usando la meccanica newtoniana. Il problema con questo metodo, però, rimane che il basso grado di accuratezza della sua stima dei parametri più importanti, il grado di torsione nei siti di legame. Ciò significa che anche l'accuratezza della sua stima dell'affinità di legame complessiva è scarsa.
Ora, Fujitsu Laboratories ha sviluppato una tecnologia di simulazione molecolare che stima il grado di torsione in una sostanza chimica, che è direttamente connesso all'affinità di legame prevista. La nuova tecnologia non tiene conto solo del punto di incollaggio in cui si verificherà la torsione, ma anche l'impatto degli atomi vicini. I laboratori Fujitsu hanno valutato questa tecnologia per 190 tipi di sostanze chimiche, confrontando i risultati con i risultati corretti ottenuti dal primo calcolo dei principi e quindi valutando il tasso di errore. Così facendo, è stato in grado di confermare che il tasso di errore nella stima del grado di torsione era, in media, un decimo rispetto alla tecnologia precedente. Si prevede che l'uso di questa nuova tecnologia nella scoperta di farmaci basati sull'IT, con la sua capacità di stimare con precisione l'affinità di legame di proteine mirate e sostanze chimiche, offre il potenziale per sforzi innovativi di scoperta di nuovi farmaci che non potrebbero essere raggiunti con approcci precedenti.
La scoperta di nuovi farmaci richiede spese e tempi significativi che possono essere misurati in decenni, portando a una ricerca globale di nuovi metodi per scoprire i farmaci. Uno dei metodi che ha riscosso un notevole interesse è la scoperta di farmaci basati sull'informatica, un nuovo metodo di scoperta di farmaci che utilizza computer che consente di creare sostanze chimiche come candidati per nuovi farmaci con un'alta probabilità di successo. La scoperta di farmaci basata sull'IT è diventata un punto focale per le aspettative come tecnologia innovativa per la creazione di nuovi farmaci, perché a differenza dei precedenti metodi di prova ed errore, in cui le sostanze chimiche vengono ripetutamente create e testate, questo approccio consente di progettare virtualmente le sostanze chimiche e di stimarne gli effetti.
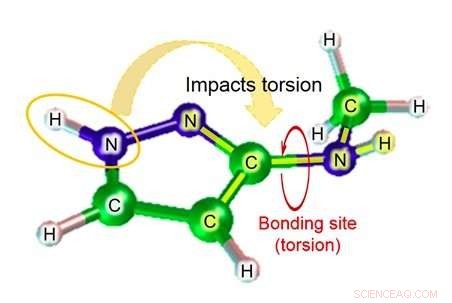
Figura 2:Esempio di struttura molecolare:3-(metilammino) pirazolo. Credito:Fujitsu
Gli effetti di una sostanza chimica come farmaco sono espressi quando la sostanza chimica si lega a una proteina bersaglio. Quando la sostanza chimica si lega alla proteina bersaglio, può cambiare la sua forma in linea con quella della proteina bersaglio. Il grado di deformazione, vale a dire, i parametri che indicano l'entità di questo cambiamento di forma, è direttamente connesso all'affinità di legame della sostanza e della proteina, e dà un'idea approssimativa del suo effetto come farmaco. Dato ciò, c'è una forte domanda per la capacità di prevedere con precisione questo valore. Per calcolare il grado di deformazione di una sostanza chimica, esistono metodi basati sulla meccanica quantistica e metodi basati sulla meccanica newtoniana. Il calcolo dei primi principi basato sulla meccanica quantistica consente calcoli estremamente accurati, risolvere gli stati degli elettroni dai tipi e dalle posizioni degli atomi coinvolti. D'altra parte, però, la capacità dei primi principi di eseguire calcoli rigorosi porta necessariamente a un enorme tempo necessario per completare i calcoli. Per simulare il grado di deformazione di numerose sostanze chimiche, il tempo necessario è dell'ordine degli anni, rendendo questo metodo impraticabile. D'altra parte, i calcoli approssimativi basati su simulazioni molecolari sono estremamente veloci, usando la meccanica newtoniana per calcolare le forze tra gli atomi all'interno delle molecole, e può anche gestire grandi molecole come le proteine abbastanza facilmente. Di conseguenza, questo metodo è ampiamente utilizzato. Con la meccanica newtoniana, le forze tra gli atomi sono espresse nel modo seguente:
Tra questi, quando una sostanza chimica è legata a una proteina bersaglio, il grado di torsione del legame rappresenta l'importante grado di deformazione. Con la tecnologia esistente, però, l'accuratezza della stima del parametro dell'angolo diedro (figura 1), che è necessario per calcolare il grado di torsione del legame, è piuttosto basso, con conseguente problema di scarsa accuratezza nella stima dell'affinità del legame nella simulazione.
Fujitsu Laboratories sviluppa tecnologie di simulazione molecolare da oltre dieci anni. Ora, utilizzando le conoscenze acquisite attraverso sforzi precedenti, Fujitsu Laboratories ha sviluppato una tecnologia di simulazione molecolare in grado di stimare il parametro dell'angolo diedro prendendo in considerazione l'impatto degli atomi vicino al legame. La tecnologia esistente stima il parametro dell'angolo diedro sulla base di un totale di quattro atomi:i due atomi nel relativo legame, e gli altri atomi a cui ciascuno di quegli atomi era legato. A seconda della struttura della molecola, però, ci sono casi in cui gli atomi oltre quei quattro potrebbero avere un impatto significativo, e in quei casi, il margine di errore della stima potrebbe essere piuttosto ampio. Con questa tecnologia, Fujitsu Laboratories ha creato un database di formule di stima per modelli di struttura parziale in cui l'impatto degli atomi più lontani dal sito di legame potrebbe essere significativo, nonché per il grado di torsione delle sostanze chimiche che ci si aspetterebbe in tal caso. Utilizzando la relativa formula di stima per trovare il grado di torsione (figura 2) nel caso di molecole corrispondenti al database per strutture parziali, è diventato possibile anche fare stime molto accurate per la torsione molecolare, che in precedenza era difficile da calcolare con precisione.
Quando Fujitsu Laboratories ha integrato questa tecnologia nel software che aveva sviluppato per generare parametri sofisticati per le forze tra gli atomi (FF-FOM), è stato in grado di confermare che i risultati erano conformi a calcoli accurati.

Figura 3:valutazione delle prestazioni dei valori dei parametri dell'angolo diedro utilizzando 190 tipi di strutture di composti chimici. Credito:Fujitsu
Quando i Fujitsu Laboratories hanno valutato la differenza tra i risultati di questa tecnologia e i risultati di un calcolo da principi primi per la stima del grado di torsione con 190 tipi di sostanze chimiche, era meno di un decimo rispetto alla tecnologia precedente, in media, 0,6 kcal/mol al di sotto delle fluttuazioni termiche della temperatura ambiente, confermando che la nuova tecnologia è pratica. Poiché può stimare con precisione l'affinità di legame delle proteine bersaglio e delle sostanze chimiche, si prevede che l'uso di questa tecnologia porterà alla creazione di nuovi farmaci pionieristici attraverso il suo utilizzo nella scoperta di farmaci basati sull'IT.