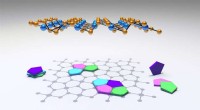
L'apprendimento automatico prevede la struttura e la dinamica delle nanoparticelle Le nanostrutture come queste nanoparticelle d'oro ricoperte di tiolo possono ora essere studiate utilizzando il nuovo metodo di apprendimento automatico sviluppato nell'Università di Jyväskylä. Il metodo può prevedere in modo affidabile l'energia potenziale di una data struttura. Credito:Antti Pihlajamäki/The University of Jyväskylä
I ricercatori del Nanoscience Center e della Facoltà di tecnologia dell'informazione dell'Università di Jyväskylä in Finlandia hanno dimostrato che i nuovi metodi di apprendimento automatico a distanza sviluppati presso l'Università di Jyväskylä sono in grado di prevedere in modo affidabile le strutture e la dinamica atomica delle nanoparticelle. I nuovi metodi sono significativamente più veloci dei metodi di simulazione tradizionali utilizzati per la ricerca sulle nanoparticelle e faciliteranno esplorazioni più efficienti delle reazioni particella-particella e della funzionalità delle particelle nel loro ambiente. Lo studio è stato pubblicato in un numero speciale dedicato al machine learning nel Giornale di chimica fisica il 15 maggio 2020.
I nuovi metodi sono stati applicati a nanoparticelle metalliche stabilizzate con ligando, che sono stati a lungo studiati al Nanoscience Center dell'Università di Jyväskylä. L'anno scorso, i ricercatori hanno pubblicato un metodo in grado di prevedere con successo i siti di legame delle molecole di ligandi stabilizzanti sulla superficie delle nanoparticelle. Ora, è stato creato un nuovo strumento in grado di prevedere in modo affidabile l'energia potenziale in base alla struttura atomica della particella, senza la necessità di utilizzare calcoli di struttura elettronica numericamente pesanti. Lo strumento facilita le simulazioni Monte Carlo della dinamica atomica delle particelle a temperature elevate.
L'energia potenziale di un sistema è una quantità fondamentale nella nanoscienza computazionale, poiché consente valutazioni quantitative della stabilità del sistema, velocità delle reazioni chimiche e forza dei legami interatomici. Le nanoparticelle metalliche stabilizzate con ligando hanno molti tipi di legami interatomici di diversa forza chimica, e tradizionalmente le valutazioni energetiche sono state fatte utilizzando la cosiddetta teoria del funzionale della densità (DFT) che spesso si traduce in calcoli numericamente pesanti che richiedono l'uso di supercomputer. Ciò ha precluso simulazioni efficienti per comprendere le funzionalità delle nanoparticelle, per esempio., come catalizzatori, o interazioni con oggetti biologici come proteine, virus, o DNA. Metodi di apprendimento automatico, una volta addestrato a modellare i sistemi in modo affidabile, può velocizzare le simulazioni di diversi ordini di grandezza.
Il nuovo metodo ha permesso di eseguire le simulazioni su un laptop o desktop
In questo lavoro i ricercatori hanno utilizzato le energie potenziali, previsto dal metodo di apprendimento automatico, per simulare la dinamica atomica di nanoparticelle d'oro stabilizzate con tiolo. I risultati erano in buon accordo con le simulazioni eseguite utilizzando la teoria del funzionale della densità. Il nuovo metodo ha consentito di eseguire simulazioni su un laptop o desktop in un arco di tempo di poche ore, mentre le simulazioni DFT di riferimento richiedevano giorni in un supercomputer e utilizzavano contemporaneamente centinaia o addirittura migliaia di core di computer. L'accelerazione consentirà simulazioni a lungo termine dei cambiamenti strutturali delle particelle e delle reazioni particella-particella a temperature elevate.
I ricercatori hanno utilizzato un metodo di apprendimento automatico a distanza sviluppato nel gruppo del professor Tommi Kärkkäinen a Jyväskylä. Descrive ogni configurazione atomica momentanea di una nanoparticella calcolando un cosiddetto descrittore, e confronta le distanze tra i descrittori in uno spazio numerico multidimensionale. Utilizzando le correlazioni a un training set creato dalle simulazioni DFT di riferimento, l'energia potenziale può essere prevista. Questo approccio, utilizzato ora per la prima volta nella ricerca sulle nanoparticelle, è più semplice e trasparente rispetto alle reti neurali tradizionalmente utilizzate.
"È estremamente motivante poter ridurre il carico computazionale dall'esecuzione di simulazioni nei supercomputer all'esecuzione con qualità simile in un laptop o un PC di casa, " dice il dottorando Antti Pihlajamäki che è l'autore principale dello studio.
"È stata una grande sorpresa che i nostri metodi di apprendimento automatico relativamente semplici funzionino così bene per nanostrutture complicate, " afferma il professor Tommi Kärkkäinen.
"Nella fase successiva, il nostro obiettivo è generalizzare il metodo affinché funzioni bene per nanoparticelle di molte dimensioni e composizioni chimiche diverse. Avremo ancora bisogno di supercomputer per generare dati di alta qualità sufficienti per addestrare l'algoritmo di apprendimento automatico, ma speriamo che in futuro possiamo passare a utilizzare questi nuovi metodi principalmente per studi di funzionalità delle nanoparticelle in ambienti chimici complicati, " riassume il professore dell'Accademia Hannu Häkkinen, che ha coordinato lo studio.