Gli scienziati hanno utilizzato Diamond Light Source per sviluppare un nuovo metodo per estrarre informazioni precedentemente nascoste dai dati di diffrazione dei raggi X che vengono misurati durante la risoluzione delle strutture atomiche tridimensionali (3D) di proteine e altre molecole biologiche.
Quando si cerca di evolvere composti chimici verso potenti candidati farmaci, gli scienziati tentano di studiare i dettagli atomici di come i composti si legano alle loro proteine bersaglio. Fare così, confrontano i dati dei raggi X misurati sia in presenza che in assenza del composto. Però, con algoritmi di analisi esistenti, questo segnale di differenza può spesso essere sommerso dal rumore degli artefatti dell'esperimento, rendendo molto inaffidabile interpretare il segnale osservato.
Il nuovo metodo Pan-Dataset Density Analysis (PanDDA) estrae l'immagine del composto legato con dettagli eccezionalmente chiari e non ambigui. PanDDA identifica prima la fonte del rumore, e poi lo rimuove dai dati. Sfrutta la capacità di Diamond di ripetere rapidamente da decine a centinaia di misurazioni, che poi si caratterizzano per le differenze tra loro, indicando la presenza di composto legato, dopodiché viene applicata una correzione del rumore in 3D. I risultati sono pubblicati oggi in Comunicazioni sulla natura .
Cristallografia macromolecolare (MX), la tecnica a cui si applica PanDDA, è uno degli strumenti più potenti utilizzati dai ricercatori interessati a determinare le strutture 3D di grandi molecole biologiche, comprese le proteine, ed è l'esperimento del cavallo di battaglia per la progettazione razionale di farmaci.
"Il problema di identificare gli eventi vincolanti nei set di dati cristallografici può sembrare come cercare un ago in un pagliaio, " spiega il dottor Nicholas Pearce, autore principale dell'articolo che deriva dal suo progetto di dottorato presso l'Università di Oxford nel Centro di formazione per dottorati (SABS) Systems Approaches for Biomedical Science, dove è stato finanziato congiuntamente da UCB Pharma e Diamond. "Nel caso dei dati che stavamo analizzando, era anche peggio, perché avevamo centinaia di mucchi di fieno, e non sapevo quale di loro contenesse aghi." Nick ora lavora nel Crystal &Structural Chemistry Group presso l'Universiteit Utrecht.
I ricercatori sono stati in grado di sfruttare a loro vantaggio il fatto che la maggior parte delle misurazioni provenivano da cristalli "vuoti" che non contenevano un ligando legato, consentendo loro di caratterizzare la forma non associata e semplicemente di cercare set di dati diversi.
"Spesso in cristallografia puoi perdere le forme 'deboli' legate, perché ogni misura è una sovrapposizione delle forme legate e non legate, " continua il dottor Pearce. "Questo è simile a più fogli di carta da lucido, ciascuno con una di almeno due immagini, tutti sovrapposti l'uno sull'altro."
"Quando si cerca di identificare l'immagine su uno solo dei 'fogli', si confonde con ciò che traspare da tutti gli altri fogli, quindi l'immagine diventa suscettibile di errori di interpretazione, "Il dottor Pearce aggiunge. "Per superare questo, abbiamo sviluppato un metodo per estrarre la giusta serie di 'fogli' dalla sovrapposizione; una volta che l'abbiamo fatto, l'interpretazione della forma rilegata diventa molto più semplice, e ci consente di interpretare con sicurezza i dati, e costruire modelli degli stati interessanti nei dati."
"L'idea di base è concettualmente molto semplice, vale a dire trattare la sovrapposizione confusa come un problema di correzione del fondo, " spiega il professor Frank von Delft, che è insieme Principal Investigator del gruppo Protein Crystallography nello Structural Genomics Consortium (SGC) presso l'Università di Oxford, e Principal Beamline Scientist della linea di luce I04-1 a Diamond. "Però, una stima accurata dello sfondo è fondamentale, e in pratica questo era impensabile fino all'avvento della nuova tecnologia robotica offerta da Diamond, il che rende di routine eseguire un numero così elevato di misurazioni."
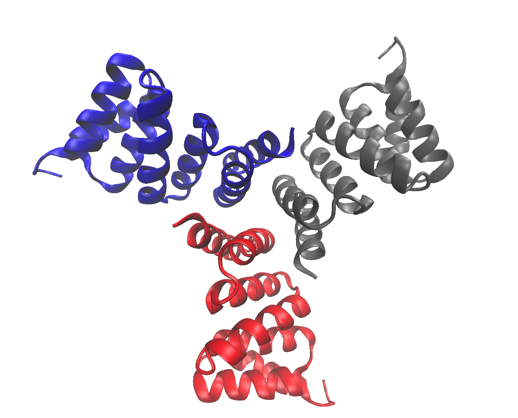
"UCB è lieta di aver lavorato a stretto contatto con Diamond per lo sviluppo di PanDDA e la sua applicazione allo screening dei frammenti cristallografici, " commenta il dottor Neil Weir, Vicepresidente senior di Discovery presso UCB Pharma. "Come diretta conseguenza, siamo stati in grado di identificare frammenti, che altrimenti non sarebbero distinguibili dallo sfondo, legato a un bersaglio farmacologico chiave per l'interazione proteina-proteina".
La ricerca ha comportato la produzione di circa 860 set di dati, di cui solo 75 contengono una forma vincolata di interesse per i ricercatori. "Sebbene generale applicabile in MX, il metodo è particolarmente trasformativo per una versione dell'esperimento MX chiamata screening dei frammenti, dove gli effetti che stiamo cercando sono molto rari e ancora più difficili da verificare con algoritmi convenzionali, "continua von Delft.
Una coda cruciale per il lavoro è stato il caricamento di tutte le strutture nel Protein Data Bank (wwPDB), il repository online di strutture 3D di proteine e acidi nucleici, dove tutti hanno libero accesso a tutte le strutture mai pubblicate. Uno dei siti host di wwPDB, RCSB PDB, ha recentemente sviluppato un nuovo strumento di deposizione di gruppo per consentire il caricamento di massa di strutture, e questo è stato fondamentale per completare questa collaborazione.
Il sistema RCSB PDB Group Deposition consente agli autori di sfruttare i modelli locali e PDB_extract per l'elaborazione batch, packing, upload, review, validation, and one-click submission of many structures at once. Searching group title "PanDDA analysis group deposition" at rcsb.org will return these 860 depositions.
"The Diamond and PDB groups have accomplished something quite incredible, and we have been delighted to help them" says Aled Edwards, Director of the SGC. "I would also like to highlight the team's commitment to open science. By placing all the research output into the public domain, they have ensured that the data can be used by all."
Now celebrating its 10th year of research and innovation, Diamond is committed to working with our users to enable them to carry out world-leading research at the facility.
"We've come a long way in the last ten years, and collaborations like these are key to how we will maintain our place as a key facility for researchers working in the life sciences, " adds Professor Dave Stuart, Director of Life Sciences at Diamond. "The idea that we can clearly see weak binding events is particularly exciting and something we're looking forward to sharing with our crystallography community."
The researchers hope that this new method will provide a significant shift in how crystallographic models are generated; opening windows to explore more poorly ordered crystals.