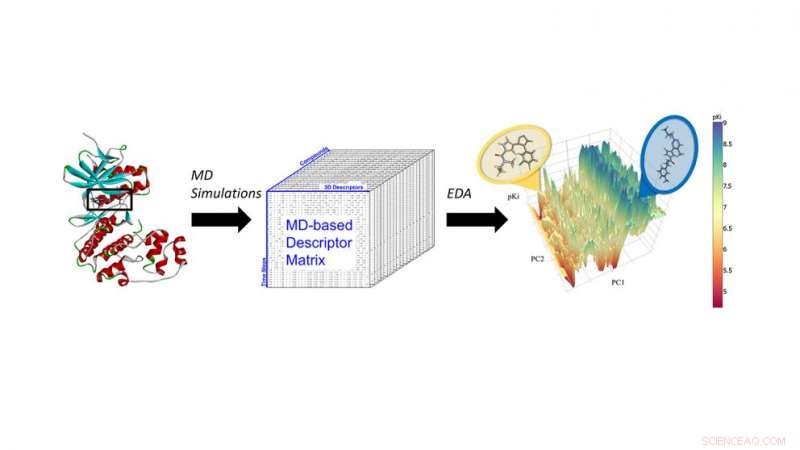
Simulazioni di dinamica molecolare (MD) di inibitori ERK2 per estrarre descrittori MD per l'analisi chemininformatica di nuova generazione e l'apprendimento automatico. Credito:North Carolina State University
I ricercatori della North Carolina State University hanno dimostrato che le simulazioni di dinamica molecolare e le tecniche di apprendimento automatico potrebbero essere integrate per creare modelli di previsione al computer più accurati. Questi modelli "iper-predittivi" potrebbero essere utilizzati per prevedere rapidamente quali nuovi composti chimici potrebbero essere promettenti candidati a farmaci.
Lo sviluppo di farmaci è un processo costoso e dispendioso in termini di tempo. Per restringere il numero di composti chimici che potrebbero essere potenziali candidati a farmaci, gli scienziati utilizzano modelli informatici in grado di prevedere come un particolare composto chimico potrebbe interagire con un bersaglio biologico di interesse, ad esempio, una proteina chiave che potrebbe essere coinvolta in un processo patologico. Tradizionalmente, questo viene fatto tramite la modellazione quantitativa della relazione struttura-attività (QSAR) e l'aggancio molecolare, che si basano su informazioni bidimensionali e tridimensionali su tali sostanze chimiche.
Denis Fourches, professore assistente di chimica computazionale, voleva migliorare la precisione di questi modelli QSAR. "Quando esamini una serie di 30 milioni di composti, non hai necessariamente bisogno di un'affidabilità molto elevata con il tuo modello:ti stai solo facendo un'idea del 5 o 10 percento superiore di quella libreria virtuale. Ma se stai cercando di restringere un campo di 200 analoghi a 10, che è più comunemente il caso nello sviluppo di farmaci, la tua tecnica di modellazione deve essere estremamente accurata. Le tecniche attuali non sono sicuramente abbastanza affidabili".
Fourches e Jeremy Ash, uno studente laureato in bioinformatica, ha deciso di incorporare i risultati dei calcoli di dinamica molecolare - simulazioni di tutti gli atomi di come un particolare composto si muove nella tasca di legame di una proteina - in modelli di previsione basati sull'apprendimento automatico.
"La maggior parte dei modelli utilizza solo le strutture bidimensionali delle molecole, " dice Fourches. "Ma in realtà, le sostanze chimiche sono oggetti tridimensionali complessi che si muovono, vibrano e hanno interazioni intermolecolari dinamiche con la proteina una volta ancorata nel suo sito di legame. Non puoi vederlo se guardi solo la struttura 2-D o 3-D di una data molecola".
In uno studio di prova del concetto, Fourches e Ash hanno esaminato la chinasi ERK2 - un enzima associato a diversi tipi di cancro - e un gruppo di 87 noti inibitori di ERK2, che vanno da molto attivo a inattivo. Hanno eseguito simulazioni di dinamica molecolare (MD) indipendenti per ciascuno di questi 87 composti e calcolato informazioni critiche sulla flessibilità di ciascun composto una volta nella tasca ERK2. Quindi hanno analizzato i descrittori MD utilizzando tecniche di chemininformatica e apprendimento automatico. I descrittori MD sono stati in grado di distinguere con precisione gli inibitori ERK2 attivi da attivi e inattivi debolmente, il che non era il caso quando i modelli utilizzavano solo informazioni strutturali 2-D e 3-D.
"Avevamo già dati su queste 87 molecole e sulla loro attività all'ERK2, " Fourches dice. "Così abbiamo testato per vedere se il nostro modello era in grado di trovare in modo affidabile i composti più attivi. Infatti, ha accuratamente distinto tra inibitori ERK2 forti e deboli, e poiché i descrittori MD codificano le interazioni che quei composti creano nella tasca di ERK2, ci ha anche fornito maggiori informazioni sul motivo per cui i potenti inibitori hanno funzionato bene.
"Prima che i progressi informatici ci permettessero di simulare questo tipo di dati, ci sarebbero voluti sei mesi per simulare una singola molecola nella tasca di ERK2. Grazie all'accelerazione GPU, ora ci vogliono solo tre ore. Questo è un punto di svolta. Sono fiducioso che l'incorporazione dei dati estratti dalla dinamica molecolare nei modelli QSAR consentirà una nuova generazione di modelli iper-predittivi che contribuiranno a portare nuovi, farmaci efficaci sul mercato ancora più velocemente. È l'intelligenza artificiale che lavora per noi per scoprire le droghe di domani".