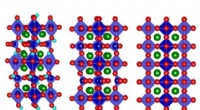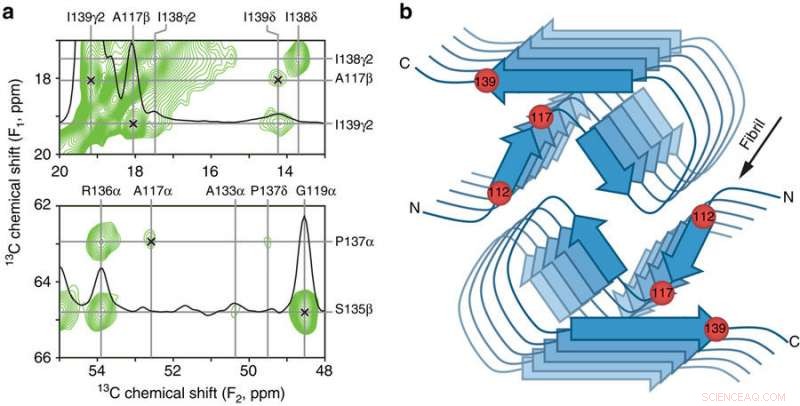
Contatti interresidui chiave e modello schematico del nucleo amiloide PrP23-144 umano. a Piccole regioni di uno spettro NMR a stato solido 13C-13C DARR bidimensionale da 900 MHz registrato con un tempo di miscelazione di 500 ms per le fibrille amiloidi generate da huPrP23-144 espresse con piruvato 3-13C come fonte di carbonio. Le regioni spettrali contengono i vincoli chiave sulla struttura del nucleo dell'amiloide [hu] sotto forma di correlazioni non ambigue a lungo raggio (indicate da segni x) tra i seguenti atomi di 13C:A117Cβ-I139Cγ2, A117Cβ-I139Cδ, A117Cα-P137Cα, e G119Cα-S135Cβ. b Modello schematico per il nucleo amiloide [hu] basato sulla combinazione di NMR a stato solido e dati di microscopia elettronica a trasmissione a fascio inclinato (vedere il testo per i dettagli). In questo modello [hu] le fibrille amiloidi sono costituite da due protofilamenti in una disposizione simmetrica C2 con regioni -sheet che corrono parallele all'asse lungo delle fibrille. Le posizioni approssimative dei residui di amminoacidi 112, 117, e 139, che hanno un impatto maggiore sulla struttura adottata dall'amiloide PrP23-144 come discusso nel testo, sono indicati da sfere rosse. Credito: Comunicazioni sulla natura (2017). DOI:10.1038/s41467-017-00794-z
I ricercatori che studiano una proteina che causa una malattia degenerativa ereditaria del cervello negli esseri umani hanno scoperto che l'essere umano, forme di topo e criceto della proteina, che hanno sequenze di amminoacidi quasi identiche, mostrano strutture tridimensionali distinte a livello atomico.
La proteina provoca angiopatia amiloide cerebrale umana familiare (CAA), e lo studio, che appare in Comunicazioni sulla natura , è il primo ad esaminare le forme della proteina in tre specie diverse.
Christopher Jaronic, professore di chimica e biochimica alla Ohio State University, ha affermato che i risultati evidenziano il fatto che alterazioni minori nei singoli amminoacidi possono causare profonde differenze nella struttura e nella funzione tra questa famiglia di proteine.
"Le differenze su larga scala nelle strutture e nelle caratteristiche di trasmissione di queste proteine, causate da differenze apparentemente insignificanti nelle posizioni di alcuni atomi di carbonio e di idrogeno, sono piuttosto notevoli, " disse Jaronic.
Lo studio non costituisce la base per un nuovo test o trattamento per CAA, ma usa piuttosto queste proteine come modelli per comprendere gli aspetti fondamentali della trasmissione tra specie di un'intera classe di malattie degenerative del cervello note come malattie da prioni, Lui ha spiegato. Sottolinea inoltre l'utilità della spettroscopia di risonanza magnetica nucleare (NMR) allo stato solido per l'imaging delle strutture delle proteine associate alle malattie da prioni.
I ricercatori sanno che nel corpo, le molecole proteiche associate alla CAA formano placche che si depositano nelle pareti dei vasi sanguigni nel cervello, ma non ci sono stati esami dettagliati della struttura molecolare di queste placche fino a tempi recenti. Nel 2008, I ricercatori dello Stato dell'Ohio e i loro partner della Case Western Reserve University hanno eseguito gli studi iniziali a stato solido della variante della proteina prionica pertinente, e ha ristretto l'elenco degli amminoacidi possibilmente critici per la sua funzione a circa 30.
Ora, hanno dimostrato che un singolo amminoacido, noto per il suo numero lungo la catena proteica, 139 - è la chiave per questa variante della proteina prionica che adotta una struttura "simile a quella umana" rispetto a una "simile a un criceto", mentre un altro amminoacido, 112, governa le differenze strutturali tra la versione umana e quella murina della proteina. Hanno anche dimostrato che questi due amminoacidi sembrano essere responsabili dell'emergere di "ceppi di prioni" strutturalmente distinti all'interno della stessa sequenza proteica, analogamente a ceppi distinti di un virus.
Le malattie da prioni più note includono l'encefalopatia spongiforme bovina (spesso chiamata "malattia della mucca pazza") e la malattia di Creutzfeldt-Jakob nell'uomo. Tutti sono incurabili e fatali, e alcuni possono anche essere trasmissibili. Si ritiene che le strutture adottate dalle proteine prioniche del cervello all'interno delle placche siano fondamentali per la loro capacità di essere trasmesse tra diversi ospiti e causare malattie.
"Il nostro gruppo sta attualmente lavorando per determinare le strutture molecolari ad alta risoluzione delle varianti della proteina prionica troncata associate alla CAA umana familiare al fine di ottenere una comprensione atomistica completa dei fattori alla base della loro trasmissione, e il presente studio è un importante trampolino di lancio in questo sforzo, " disse Jaronic.
"Speriamo che un giorno il nostro gruppo e altri ricercatori saranno in grado di utilizzare metodologie simili per svelare le basi strutturali delle malattie da prioni trasmissibili, " Ha aggiunto.