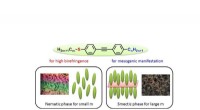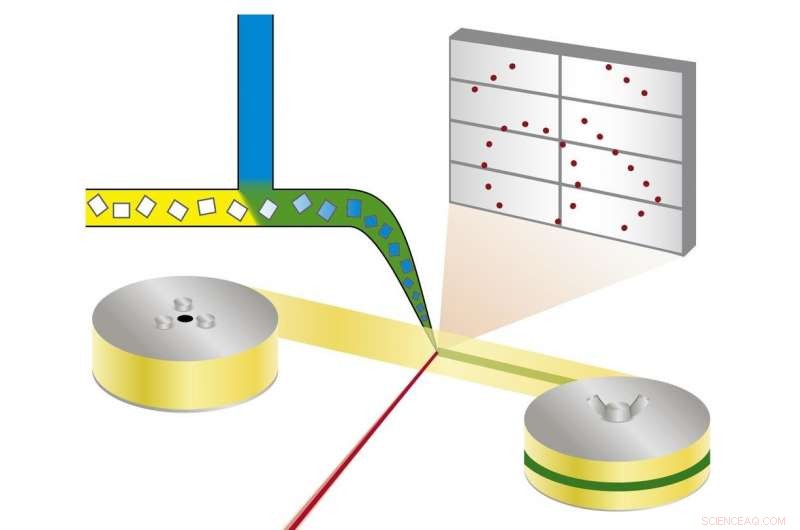
Principio della cristallografia seriale di sincrotrone mix-and-diffuse:i cristalli proteici vengono miscelati con una soluzione di un farmaco candidato e sottoposti a raggi X su un nastro che attraversa il raggio di raggi X. Credito:Beyerlein et al., IUCrJ
Gli scienziati di DESY hanno sviluppato un nuovo metodo che consente uno screening rapido e automatizzato di promettenti farmaci candidati. Questa nuova tecnica, chiamato cristallografia di sincrotrone seriale mix-and-diffuse, può immaginare l'interazione di potenziali bersagli farmacologici con farmaci candidati o altre molecole. Il concetto ha il potenziale per portare la progettazione di farmaci basata su struttura e frammento a un nuovo livello, come scrivono i ricercatori nel Giornale dell'Unione Internazionale di Cristallografia ( IUCrJ ).
Molte proteine nel corpo sono potenziali bersagli farmacologici. Le molecole farmaceutiche con la forma corretta possono legarsi a queste proteine e attivare o disattivare la loro funzione. Ad esempio, per combattere alcune forme di leucemia, il farmaco antitumorale Imatinib inibisce una variante iperattiva dell'enzima tirosin-chinasi, una proteina responsabile dell'attivazione di molte altre proteine. Imatinib blocca il sito attivo di questa tirosin-chinasi. Per realizzare questo, la molecola del farmaco deve inserirsi con precisione nel sito attivo come una chiave in una serratura. Sulla base della conoscenza della struttura spaziale del bersaglio enzimatico, Imatinib è stato creato su misura per questo scopo.
"Questa strategia è chiamata progettazione di farmaci basata sulla struttura ed è oggi utilizzata come metodo standard nello sviluppo di farmaci farmaceutici, " spiega il primo autore Kenneth Beyerlein del Center for Free-Electron Laser Science (CFEL), una collaborazione di DESY, l'Università di Amburgo e la società tedesca Max Planck. "Però, in realtà prendere di mira le proteine è molto più complesso che inserire una chiave in una serratura. Perciò, molte potenziali molecole farmaceutiche o frammenti di tali molecole devono essere testate, che di solito è una procedura lunga e complicata." Inoltre, tanto i biologi quanto i farmacologi sono interessati al preciso funzionamento degli agenti naturali che si legano alle proteine, per capire meglio il meccanismo della vita.
Il sistema sviluppato dal team di Beyerlein e dal suo collega DESY Dominik Oberthür, anche da CFEL, offre un nuovo modo di perseguire questo obiettivo:mescola proteine microcristalline con molecole specifiche chiamate ligandi che possono essere candidati a farmaci o agenti naturali appena prima di sondare i cristalli con i raggi X per rivelare la struttura spaziale dettagliata del complesso proteina-ligando risultante o il assenza di tale complesso se un potenziale ligando non si lega alla proteina.
Per analizzare la struttura spaziale di una proteina, gli scienziati usano spesso la cristallografia a raggi X. Per questa tecnica, un cristallo deve essere cresciuto prima dalla proteina. I ricercatori scattano quindi istantanee a raggi X da tutti i lati del cristallo che deve essere raffreddato a temperature ultra basse per ridurre i danni causati dalle radiazioni intense. I raggi X producono un caratteristico pattern di diffrazione da cui è possibile calcolare la struttura interna del cristallo e quindi la struttura spaziale della proteina. Per studiare una proteina con un ligando, un nuovo cristallo deve essere cresciuto da una soluzione proteica e ligando o il cristallo deve essere impregnato con il ligando. Anche con l'uso della robotica per automatizzare tutte le fasi di questo processo, la necessità di montare singoli cristalli per ogni nuovo set di dati è diventata la fase limitante della velocità nello screening di grandi librerie di composti.
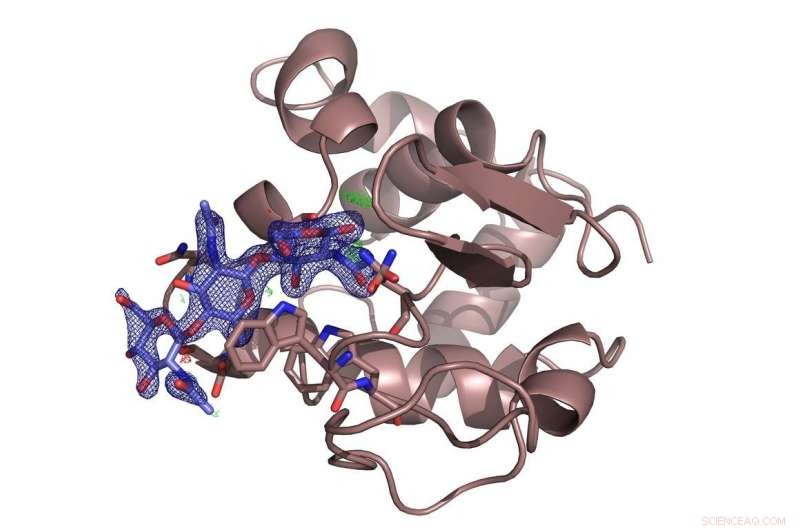
L'enzima lisozima (marrone) con lo zucchero inibitore chitotriosio (blu) legato ad esso. L'indagine ha risolto una controversia sul sito di legame preferito della molecola di zucchero. Credito:DESY, Dominik Oberthür
La nuova tecnica segue un approccio diverso. "Stiamo usando microcristalli che hanno due vantaggi:di solito sono molto più facili da produrre rispetto ai cristalli di grandi dimensioni, e sono abbastanza piccoli in modo che un potenziale farmaco in una soluzione possa diffondersi attraverso il cristallo e legarsi a tutte le molecole proteiche in pochi millisecondi, " spiega Oberthür. Il sistema sviluppato dal team di Oberthür e Beyerlein eroga un flusso di microcristalli in un liquido di trasporto su un nastro sottile. Come un nastro trasportatore, il nastro trasporta i cristalli attraverso il fascio di raggi X, che viene sminuzzato in brevi lampi da una tenda rotante. Invece di ruotare un grande cristallo nel raggio di raggi X, molti microcristalli con orientamento casuale vengono quindi sottoposti a raggi X in modo seriale e i modelli di diffrazione di ogni scatto vengono successivamente combinati per formare un set di dati completo, seguendo il concetto di cristallografia seriale che è stato sviluppato per la prima volta con i laser a raggi X a elettroni liberi (XFEL).
Attraverso una seconda valvola nel sistema, viene aggiunta una soluzione di un candidato farmaco o di un ligando naturale. Il punto in cui i due liquidi si mescolano può essere regolato per creare un ritardo definito prima di indagare sulla struttura. Questa configurazione non richiede il crio-raffreddamento dei cristalli, quindi l'interazione proteina-farmaco può essere osservata a temperature fisiologiche, o qualsiasi altra temperatura desiderata. Per di qua, anche le dinamiche di legame possono essere studiate. "Possiamo diffondere al volo sostanze chimiche nei cristalli proteici e osservare il legame che avviene, " spiega Oberthür. "Non è necessario trovare nuove condizioni di crescita per ciascun inibitore e non è necessario scambiare i cristalli manualmente, l'intero processo può essere automatizzato."
Il team ha testato il nuovo sistema presso la sorgente di raggi X ad alta brillantezza PETRA III di DESY con il noto lisozima proteico e una molecola di zucchero, chitotriosio, che inibisce l'enzima. I microcristalli di lisozima usati qui avevano un diametro di soli sei-otto micrometri. L'allestimento presso la stazione di misurazione P11 ha rivelato in dettaglio la struttura spaziale dell'inibitore misto legato al lisozima. E anche se la struttura del lisozima è stata la prima struttura enzimatica rivelata dalla cristallografia a raggi X 50 anni fa, il nuovo metodo potrebbe ancora rivelare nuovi dettagli sulla modalità di legame del chitotriosio al lisozima, risolvere una controversia sul sito di legame preferito della molecola di zucchero.
Mentre la prova di principio richiedeva ancora del tempo, routine e ulteriori progressi nella tecnologia dei rivelatori e dei raggi X accelereranno notevolmente la procedura. Anche, utilizzando l'intero spettro del fascio di raggi X dalla sorgente di luce di sincrotrone invece di un solo "colore" da esso, può spingere il tempo di esposizione per le singole immagini di diffrazione fino a 100 picosecondi, o 0,1 miliardesimi di secondo. Solo 50 di queste immagini sono sufficienti per determinare la struttura, come è stato recentemente mostrato.
"Stiamo sviluppando modi per risolvere la struttura delle proteine legate per la scoperta di farmaci ad alto rendimento, " ha spiegato Beyerlein. Poiché le sorgenti di luce di sincrotrone sono più accessibili dei laser a raggi X, i ricercatori immaginano di utilizzare questo metodo per lo screening di routine attraverso librerie di potenziali inibitori e frammenti di farmaci. "Farlo automaticamente e molto più velocemente rispetto agli approcci convenzionali sarebbe un grande passo avanti nella progettazione di farmaci basata sulla struttura, "dice Beyerlein.