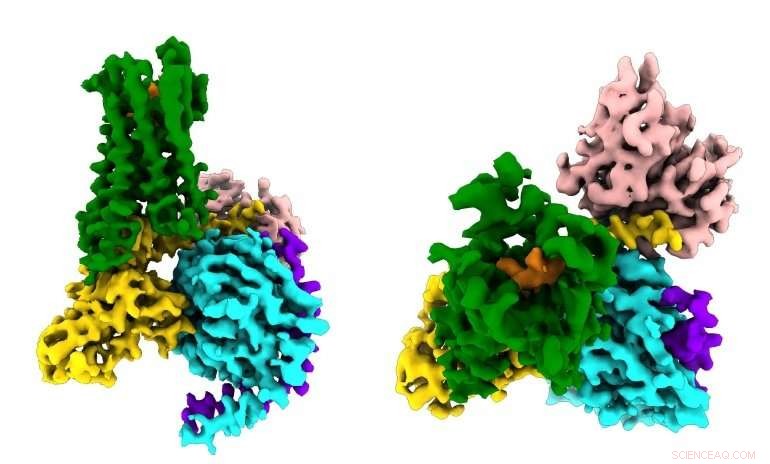
I ricercatori vedono per la prima volta come un farmaco oppioide sintetico (arancione) si lega ai recettori µ-oppioidi (verde) nel cervello, e attivare molecole di segnalazione all'interno dei neuroni (G⍺ in oro, Gβ in ciano, Gγ in viola) che portano alla soppressione del dolore e alla dipendenza. Credito:Antoine Koehl (laboratorio Manglik)
I farmaci oppioidi come la morfina e il fentanil sono un pilastro della moderna medicina del dolore. Ma causano anche stitichezza, creano una forte dipendenza, e può portare a insufficienza respiratoria fatale se assunto a dosi troppo elevate. Gli scienziati hanno a lungo cercato di sviluppare nuovi farmaci oppioidi in grado di allontanare il dolore senza questi pericolosi effetti collaterali, ma i buchi nella nostra comprensione di come esattamente gli oppioidi esercitino i loro vari effetti a livello biologico hanno finora tenuto a bada questo sogno.
Gli antidolorifici oppioidi agiscono legandosi a una proteina recettore presente sulle cellule nervose chiamata recettore µ-oppioidi, che si è evoluto per rispondere agli antidolorifici naturali del corpo (come le endorfine prodotte dall'esercizio) attenuando il dolore e creando un senso di euforia. I farmaci oppioidi, dall'oppio alla morfina, all'eroina, dirottano questo sistema di segnalazione legandosi alla stessa molecola recettore. Ma i dettagli su come l'attivazione di questi recettori innesca gli effetti positivi e negativi dei farmaci sono rimasti confusi.
Ora, in uno studio pubblicato il 13 giugno, 2018 in Natura , gli scienziati della UC San Francisco e della Stanford University hanno utilizzato la microscopia crioelettronica ad altissima risoluzione (cryoEM) per catturare il ritratto più dettagliato di sempre di un farmaco oppioide che innesca la cascata di segnali biochimici che gli conferisce il suo potere, sia nel bene che nel male .
"Abbiamo essenzialmente catturato questo evento di segnalazione nell'atto, ", ha affermato il co-autore senior dello studio Aashish Manglik, dottore, dottorato di ricerca, un assistente professore di chimica farmaceutica presso la School of Pharmacy della UCSF che ha condotto il nuovo studio come studente laureato e Distinguished Fellow a Stanford. "Si spera che queste nuove immagini a livello atomico ci consentano di progettare razionalmente composti che colpiscano diversi aspetti della segnalazione degli oppioidi nel cervello, con la speranza di identificare nuovi, antidolorifici più sicuri”.
Il recettore µ-oppioide fa parte di una vasta famiglia di centinaia di proteine di segnalazione chiamate recettori accoppiati a proteine G (GPCR) che sono coinvolti in tutto, dalla vista e dall'udito alla risposta del sistema immunitario agli agenti patogeni invasivi, e sono gli obiettivi di oltre il 30% dei farmaci moderni. La maggior parte dei GPCR condivide gli stessi meccanismi di base:quando la giusta molecola di segnalazione (ad esempio, un oppioide) si lega a un GPCR all'esterno della cellula, la proteina stimola una reazione a catena di segnali biochimici all'interno della cellula attivando una molecola messaggera chiamata proteina G (da cui il nome GPCR).
Esperimenti che hanno rivelato come un diverso tipo di GPCR si leghi alla proteina G "stimolante" ha portato a un premio Nobel per Brian Kobilka di Stanford, dottore, uno degli autori senior del nuovo studio, ma i ricercatori sanno da decenni che i GPCR possono anche legarsi a una dozzina di altre molecole di segnalazione all'interno della cellula. Per esempio, I recettori µ-oppioidi in genere attivano solo le cosiddette proteine G "inibitrici", che hanno l'effetto opposto della cascata di proteine G stimolatorie. Però, gli scienziati non sono sicuri di cosa causi l'affinità di alcuni GPCR per particolari proteine partner all'interno della cellula, o esattamente quali sono le conseguenze.
I ricercatori sperano che, comprendendo questi diversi percorsi di segnalazione GPCR, possono essere in grado di sviluppare farmaci con effetti altamente specifici, come sopprimere il dolore senza causare dipendenza. Ma fino ad ora, i ricercatori non avevano idea di come un dato GPCR interagisse selettivamente solo con un sottoinsieme di partner di segnalazione all'interno della cellula.
Il nuovo studio, pubblicato il 13 giugno 2018 in Natura , catturato per la prima volta come il recettore µ-oppioide si lega al suo partner inibitorio della proteina G. Tra gli altri ritrovamenti, lo studio ha mostrato che la selettività del recettore sembra essere dovuta alle piccole dimensioni della tasca di legame per la proteina G all'interno della cellula, mentre la proteina G stimolante richiede un sito di legame più grande.
Manglik ha precedentemente collaborato con il laboratorio di scoperta di farmaci computazionali di Brian Shoichet, dottorato di ricerca, un professore di chimica farmaceutica nella School of Pharmacy dell'UCSF, identificare una molecola chiamata PZM21 che consente al recettore µ-oppioide di impegnare solo la proteina G inibitoria ma non un'altra molecola di segnalazione chiamata beta-arrestina, e ha dimostrato che questo farmaco selettivo ha fornito sollievo dal dolore con effetti collaterali ridotti nei topi. Il suo laboratorio sta ora costruendo il nuovo, ritratto ad alta risoluzione del recettore degli oppioidi - complesso di proteine G per sviluppare nuovi, composti ancora più selettivi.