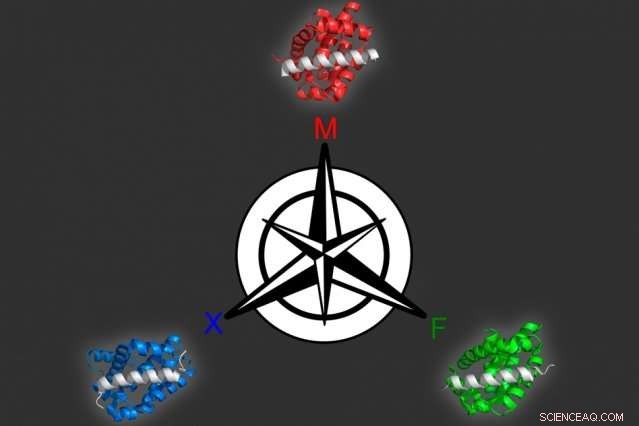
Utilizzando un approccio di modellazione al computer che hanno sviluppato, I biologi del MIT hanno identificato tre diverse proteine che possono legarsi selettivamente a ciascuno dei tre bersagli simili, tutti i membri della famiglia di proteine Bcl-2. Credito:Vincent Xue
Progettare proteine sintetiche che possono agire come farmaci per il cancro o altre malattie può essere un processo noioso:generalmente comporta la creazione di una libreria di milioni di proteine, quindi lo screening della libreria per trovare le proteine che legano il bersaglio corretto.
I biologi del MIT hanno ora escogitato un approccio più raffinato in cui utilizzano la modellazione al computer per prevedere come diverse sequenze proteiche interagiranno con il bersaglio. Questa strategia genera un numero maggiore di candidati e offre anche un maggiore controllo su una varietà di tratti proteici, dice Amy Keating, un professore di biologia e ingegneria biologica e il capo del gruppo di ricerca.
"Il nostro metodo ti offre un campo di gioco molto più ampio in cui puoi selezionare soluzioni molto diverse l'una dall'altra e che avranno diversi punti di forza e responsabilità, " dice. "La nostra speranza è che possiamo fornire una gamma più ampia di possibili soluzioni per aumentare il rendimento di quei risultati iniziali in utili, molecole funzionali”.
In un articolo apparso su Atti dell'Accademia Nazionale delle Scienze la settimana del 15 ottobre, Keating e i suoi colleghi hanno utilizzato questo approccio per generare diversi peptidi che possono colpire diversi membri di una famiglia di proteine chiamata Bcl-2, che aiutano a guidare la crescita del cancro.
I recenti dottorandi Justin Jenson e Vincent Xue sono gli autori principali dell'articolo. Altri autori sono il postdoc Tirtha Mandal, ex tecnico di laboratorio Lindsey Stretz, e l'ex postdoc Lothar Reich.
Interazioni di modellazione
farmaci proteici, chiamati anche biofarmaci, sono una classe di farmaci in rapida crescita che promette il trattamento di un'ampia gamma di malattie. Il metodo usuale per identificare tali farmaci è lo screening di milioni di proteine, scelti casualmente o selezionati creando varianti di sequenze proteiche già dimostrate essere candidati promettenti. Ciò comporta l'ingegnerizzazione di virus o lieviti per produrre ciascuna delle proteine, poi esporli al bersaglio per vedere quali legano meglio.
"Questo è l'approccio standard:o completamente a caso, o con qualche conoscenza precedente, progettare una libreria di proteine, e poi andare a pescare in biblioteca per tirare fuori i membri più promettenti, " dice Keating.
Anche se questo metodo funziona bene, di solito produce proteine ottimizzate per un solo tratto:quanto bene si lega al bersaglio. Non consente alcun controllo su altre funzionalità che potrebbero essere utili, come i tratti che contribuiscono alla capacità di una proteina di entrare nelle cellule o la sua tendenza a provocare una risposta immunitaria.
"Non c'è un modo ovvio per fare quel genere di cose:specificare un peptide caricato positivamente, ad esempio, utilizzando lo screening della libreria di forza bruta, " dice Keating.
Un'altra caratteristica desiderabile è la capacità di identificare le proteine che si legano strettamente al loro bersaglio ma non a bersagli simili, che aiuta a garantire che i farmaci non abbiano effetti collaterali indesiderati. L'approccio standard consente ai ricercatori di farlo, ma gli esperimenti si fanno più macchinosi, Keating dice.
La nuova strategia prevede innanzitutto la creazione di un modello al computer in grado di mettere in relazione le sequenze peptidiche con la loro affinità di legame per la proteina bersaglio. Per creare questo modello, i ricercatori hanno prima scelto circa 10, 000 peptidi, ciascuno 23 amminoacidi di lunghezza e struttura elicoidale, e testato il loro legame con tre diversi membri della famiglia Bcl-2. Hanno scelto intenzionalmente alcune sequenze che già sapevano si sarebbero legate bene, più altri che sapevano non l'avrebbero fatto, quindi il modello potrebbe incorporare dati su una serie di capacità di legame.
Da questo insieme di dati, il modello può produrre un "panorama" di come ciascuna sequenza peptidica interagisce con ciascun bersaglio. I ricercatori possono quindi utilizzare il modello per prevedere come altre sequenze interagiranno con gli obiettivi, e generare peptidi che soddisfano i criteri desiderati.
Utilizzando questo modello, i ricercatori hanno prodotto 36 peptidi che si prevedeva legassero strettamente un membro della famiglia ma non gli altri due. Tutti i candidati si sono comportati molto bene quando i ricercatori li hanno testati sperimentalmente, così hanno tentato un problema più difficile:identificare le proteine che si legano a due dei membri ma non al terzo. Molte di queste proteine hanno avuto successo.
"Questo approccio rappresenta un passaggio dal porre un problema molto specifico e quindi progettare un esperimento per risolverlo, a investire del lavoro in anticipo per generare questo panorama di come la sequenza è correlata alla funzione, catturare il paesaggio in un modello, e quindi poterlo esplorare a piacimento per più proprietà, " dice Keating.
Sagar Khare, professore associato di chimica e biologia chimica alla Rutgers University, afferma che il nuovo approccio è impressionante nella sua capacità di discriminare tra obiettivi proteici strettamente correlati.
"La selettività dei farmaci è fondamentale per ridurre al minimo gli effetti fuori bersaglio, e spesso la selettività è molto difficile da codificare perché ci sono così tanti concorrenti molecolari dall'aspetto simile che legheranno anche il farmaco oltre al bersaglio previsto. Questo lavoro mostra come codificare questa selettività nel progetto stesso, "dice Khare, che non è stato coinvolto nella ricerca. "Le applicazioni nello sviluppo di peptidi terapeutici seguiranno quasi certamente".
Farmaci selettivi
I membri della famiglia delle proteine Bcl-2 svolgono un ruolo importante nella regolazione della morte cellulare programmata. La disregolazione di queste proteine può inibire la morte cellulare, aiutare i tumori a crescere senza controllo, così tante aziende farmaceutiche hanno lavorato allo sviluppo di farmaci mirati a questa famiglia di proteine. Affinché tali farmaci siano efficaci, potrebbe essere importante per loro prendere di mira solo una delle proteine, perché interromperli tutti potrebbe causare effetti collaterali dannosi nelle cellule sane.
"In molti casi, le cellule tumorali sembrano utilizzare solo uno o due membri della famiglia per promuovere la sopravvivenza cellulare, " dice Keating. "In generale, è risaputo che avere un gruppo di agenti selettivi sarebbe molto meglio di uno strumento rozzo che li ha eliminati tutti."
I ricercatori hanno depositato i brevetti sui peptidi identificati in questo studio, e sperano che saranno ulteriormente testati come possibili farmaci. Il laboratorio di Keating sta ora lavorando per applicare questo nuovo approccio di modellazione ad altri obiettivi proteici. Questo tipo di modellizzazione potrebbe essere utile non solo per sviluppare potenziali farmaci, ma anche la generazione di proteine da utilizzare in applicazioni agricole o energetiche, lei dice.
Questa storia è stata ripubblicata per gentile concessione di MIT News (web.mit.edu/newsoffice/), un popolare sito che copre notizie sulla ricerca del MIT, innovazione e didattica.