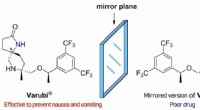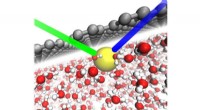
Si prevede che una libreria virtuale di molecole make-on-demand disponibili per la scoperta di farmaci supererà il miliardo di composti entro il prossimo anno. Credito:Bryan Roth, M.D., dottorato di ricerca, della University of North Carolina (UNC) Chapel Hill, Brian Shoichet, dottorato di ricerca, e John Irwin, dottorato di ricerca, dell'Università della California di San Francisco, e colleghi.
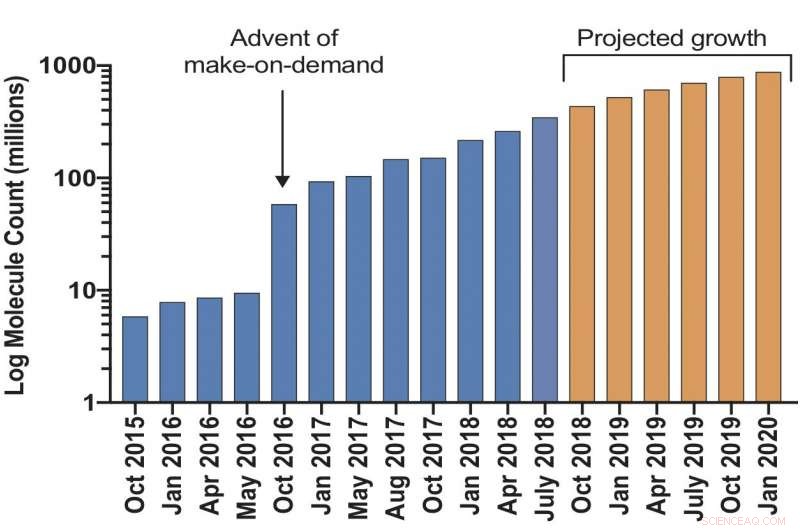
I ricercatori hanno lanciato una libreria di docking virtuale ultra-grande che dovrebbe raggiungere oltre 1 miliardo di molecole entro il prossimo anno. Espanderà di 1000 volte il numero di tali composti "make-on-demand" prontamente disponibili per gli scienziati per la biologia chimica e la scoperta di farmaci. Più grande è la biblioteca, migliori sono le sue probabilità di eliminare le molecole "esca" inattive che potrebbero altrimenti condurre i ricercatori in vicoli ciechi. Il progetto è finanziato dall'Istituto Superiore di Sanità.
"Per migliorare i farmaci per le malattie mentali, abbiamo bisogno di vagliare un numero enorme di molecole potenzialmente terapeutiche, " ha spiegato Joshua A. Gordon, M.D., dottorato di ricerca, direttore del National Institute of Mental Health (NIMH) del NIH, che ha cofinanziato la ricerca. "La modellazione computazionale imparziale ci consente di farlo in un computer, accelerando notevolmente il processo di scoperta di nuovi trattamenti. Consente ai ricercatori di "vedere" virtualmente una molecola che si aggancia alla sua proteina recettore, come una nave nel suo ormeggio o una chiave nella sua serratura, e di prevedere le sue proprietà farmacologiche, in base a come si prevede che le strutture molecolari interagiscano. Solo quelle relativamente poche molecole candidate che corrispondono meglio al profilo target sul computer devono essere fisicamente realizzate e testate in un laboratorio umido".
Bryan Roth, M.D., dottorato di ricerca, della University of North Carolina (UNC) Chapel Hill, Brian Shoichet, dottorato di ricerca, e John Irwin, dottorato di ricerca, dell'Università della California di San Francisco, e colleghi, rapporto sui loro risultati il 6 febbraio, 2019 sulla rivista Natura . Lo studio è stato sostenuto, in parte, da sovvenzioni del NIMH, Istituto Nazionale di Scienze Mediche Generali (NIGMS), il Fondo Comune NIH, e Istituto Nazionale di Malattie Neurologiche e Ictus (NINDS).
Il programma Illuminating the Druggable Genome (IDG) del NIH Common Fund, lanciato nel 2014 per catalizzare la ricerca su proteine che sono attualmente poco studiate e potenziali bersagli di intervento terapeutico, ha finanziato l'espansione della libreria di attracco.
Negli ultimi anni, Roth, Shoichet, e colleghi hanno impiegato il loro approccio di docking basato sulla struttura virtuale per scoprire i segreti molecolari di un farmaco antipsicotico e dell'LSD ancorati nei rispettivi recettori bersaglio e per creare un antidolorifico di design che si rivolge selettivamente ai circuiti analgesici del cervello senza gli effetti collaterali della morfina.
È noto l'esistenza di un numero impressionante di potenziali molecole simili a farmaci. Ancora, da centinaia di milioni a miliardi di molecole diverse sono rimaste inaccessibili a causa delle limitazioni dei metodi esistenti utilizzati per compilare librerie molecolari, dicono i ricercatori. Per esempio, la loro tecnica di docking basata su strutture virtuali, pur promettendo, rischia di trovare molti falsi positivi o "esca:i difetti nel modello consentono molecole che sembrano plausibili ma si rivelano biologicamente inattive.
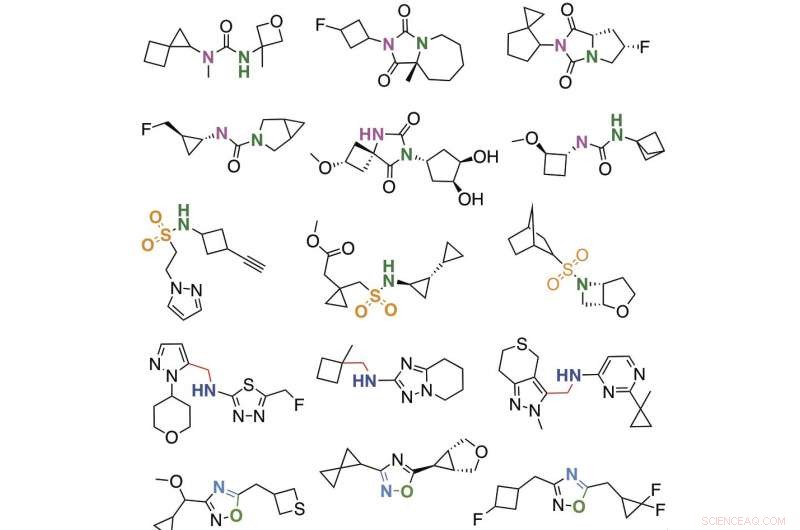
Selezione di molecole scoperte utilizzando la libreria mega docking. Credito:Bryan Roth, M.D., dottorato di ricerca, della University of North Carolina (UNC) Chapel Hill, Brian Shoichet, dottorato di ricerca, e John Irwin, dottorato di ricerca, dell'Università della California di San Francisco, e colleghi.
Per vincere questa sfida, i ricercatori si sono concentrati su molecole che risultano da 130 reazioni chimiche ben caratterizzate utilizzando 70, 000 diversi elementi costitutivi chimici. Le simulazioni al computer con queste molecole hanno mostrato che man mano che le dimensioni di una libreria crescevano, il rapporto tra "veri attivi" e esche aumentava, proprio come il potere statistico di uno studio aumenta con un campione più ampio.
Nel nuovo studio, i ricercatori hanno esaminato il docking basato sulla struttura di 138 milioni di molecole con il recettore D4, una proteina chiave che media le azioni del messaggero chimico del cervello dopamina, o l'enzima AmpC, che conferisce resistenza a determinati antibiotici e si è dimostrato difficile da bloccare.
"Il recettore D4 è di particolare interesse per NIMH a causa del suo ruolo nella cognizione e in altre funzioni esecutive della corteccia prefrontale del cervello che sono spesso disturbate nelle malattie mentali, "ha detto Laurie Nadler, dottorato di ricerca, della Divisione NIMH di Neuroscienze e Scienze Comportamentali di Base, responsabile del programma per la sovvenzione a sostegno dello studio sul recettore D4.
I ricercatori hanno quindi sintetizzato e testato, in un laboratorio, le prime 549 molecole che si sono virtualmente agganciate meglio al recettore D4 e 44 molecole che si sono agganciate meglio all'enzima. Questi studi hanno rivelato diverse nuove molecole simili a farmaci che si legano solo al recettore D4 (e non ai recettori della dopamina D2 o D3 strettamente correlati) e attivano o disattivano il recettore. Inoltre, una molecola (4163) è emersa come il più potente legante di AmpC di sempre. Il grado di attracco di una molecola virtuale ha previsto la sua effettiva probabilità di legarsi al recettore della dopamina D4 in un test di laboratorio.
La scoperta di nuove e potenti molecole per entrambi i bersagli ha anche confermato che le librerie ultra-grandi contengono molecole più adatte a una data struttura del recettore rispetto alle librerie più piccole e che il docking virtuale può riconoscere le molecole e prevedere il numero totale di composti attivi previsti all'interno di una libreria.
"Questo nuovo studio illustra il potenziale dello screening computazionale imparziale e del docking molecolare per scoprire nuove molecole strumento e potenziali agenti terapeutici, fornendo un percorso rapido e robusto che porterà direttamente a nuovi trattamenti farmacologici per le malattie mentali, "aggiunse Gordon.