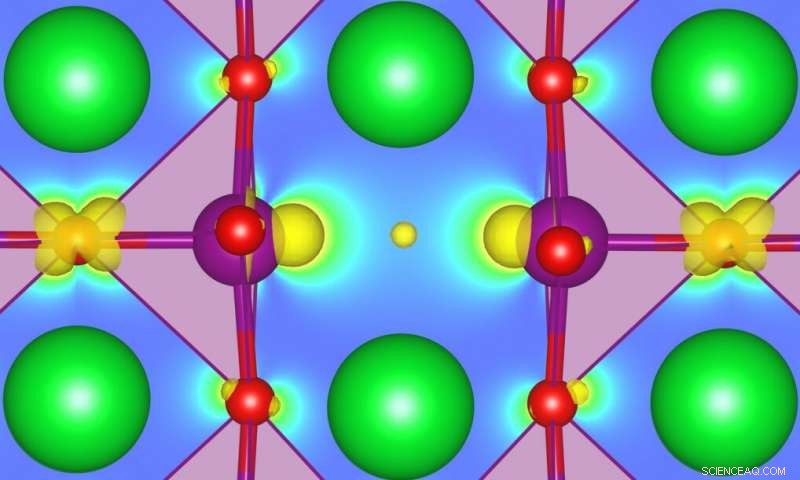
Comprendere come i difetti possono influenzare le proprietà dello stato fondamentale, promuovere le transizioni di fase, o abilitare funzionalità completamente nuove in alcuni ossidi fortemente correlati è diventato un argomento di grande interesse nel campo della progettazione e della scoperta di nuovi materiali funzionali. SrMnO3 (SMO) è un esempio particolarmente interessante, ma è necessaria una migliore caratterizzazione. I ricercatori MARVEL hanno ora sviluppato un metodo che può portare a previsioni più accurate dell'energia dei difetti associati agli stati in-gap nei semiconduttori o negli isolanti. Credito:Ulrich Aschauer
Comprendere come i difetti possono influenzare le proprietà dello stato fondamentale, promuovere le transizioni di fase, o abilitare funzionalità completamente nuove in alcuni ossidi fortemente correlati è diventato un argomento di grande interesse nel campo della progettazione e della scoperta di nuovi materiali funzionali. SrMnO 3 (SMO) è un esempio particolarmente interessante, ma è necessaria una migliore caratterizzazione. I ricercatori MARVEL hanno ora sviluppato un metodo che può portare a previsioni più accurate dell'energia dei difetti associati agli stati in-gap nei semiconduttori o negli isolanti.
Alcuni ossidi di perovskite, Per esempio, hanno mostrato un ampio spettro di proprietà funzionali tecnologicamente rilevanti come la ferroelettricità e il magnetismo che possono essere sintonizzati tramite deformazione. Sforzo, però, si accoppia anche con la chimica del difetto per determinare le proprietà del materiale.
SrMnO 3 (SMO) è un esempio particolarmente interessante per esaminare la funzionalità risultante da una complessa interazione di deformazione, ordine magnetico, distorsioni polari, e le vacanze di ossigeno che sono difetti onnipresenti in questi materiali. In particolare, la teoria ha previsto che i film sottili SMO si trasformino da antiferromagnetici a ferromagnetici con l'aumento della carenza di ossigeno, che è supportato da recenti studi sperimentali.
Queste previsioni precedenti erano tuttavia basate su calcoli della teoria del funzionale della densità (DFT) che incorporavano una correzione U basata sulle proprietà elettroniche e magnetiche delle manganiti stechiometriche. Mentre l'inclusione di U, destinata a correggere l'autointerazione degli elettroni negli ossidi complessi, è necessaria in tali materiali, la scelta specifica di U basata sulle proprietà del materiale stechiometrico potrebbe portare a potenziali carenze nella descrizione dell'SMO difettoso:gli ioni di manganese attorno al difetto hanno un ambiente di coordinazione diverso.
A seconda dello stato di carica del difetto, un ulteriore problema è legato alla descrizione di più stati di ossidazione presenti in SMO difettosi. La formazione di vacanze di ossigeno è generalmente compensata dalla carica mediante una riduzione dello stato di ossidazione (OS) degli ioni manganese adiacenti alla vacanza, che potrebbe quindi non essere propriamente descritto dallo stesso U.
Questo è il motivo per cui il postdoc dell'Università di Berna Chiara Ricca e colleghi hanno deciso che era fondamentale tenere conto degli effetti strutturali e chimici locali per ciascun sito di metallo di transizione nell'ossido quando si mirava a una descrizione accurata dell'SMO difettoso. In collaborazione con un team del laboratorio THEOS di Nicola Marzari, che ha recentemente sviluppato un approccio basato sulla teoria della perturbazione funzionale della densità (DFPT) per calcolare i parametri U, hanno usato valori U dipendenti dal sito autoconsistenti calcolati dai primi principi per studiare la chimica dei difetti e le proprietà magnetiche del bulk di SMO e dei film sottili deformati.
"Questa strettissima collaborazione tra i due gruppi, uno focalizzato sullo sviluppo di metodi e l'altro sulle applicazioni in materiali di ossido difettosi, è stato innescato dall'unione di questi diversi centri di ricerca sotto l'ombrello MARVEL", ha affermato Ulrich Aschauer dell'Università di Berna, uno dei due PI coinvolti nel lavoro.
I risultati mostrano che questa U autoconsistente migliora la struttura di SrMnO . stechiometrico 3 rispetto ad altri metodi, incluso uno che utilizza una U empirica. Per i sistemi difettosi, U cambia in funzione della distanza del sito del metallo di transizione dal difetto, il suo stato di ossidazione, il suo numero di coordinamento, e la fase magnetica del materiale. Tenendo conto di questa dipendenza, a sua volta, influenza le energie di formazione dei difetti calcolate e le transizioni di fase magnetiche previste indotte da deformazioni e/o difetti, specialmente quando gli stati localizzati occupati compaiono nella banda proibita del materiale al momento della creazione del difetto.
"Riteniamo che questo approccio possa portare a previsioni più accurate dell'energia dei difetti associati agli stati in-gap nei semiconduttori o negli isolanti sia rispetto al DFT standard che ai funzionali possibilmente ibridi a un costo computazionale significativamente inferiore rispetto a quest'ultimo, " Ha detto Ricca. "Questo grazie a una corretta descrizione degli effetti chimici strutturali e locali indotti dai difetti".