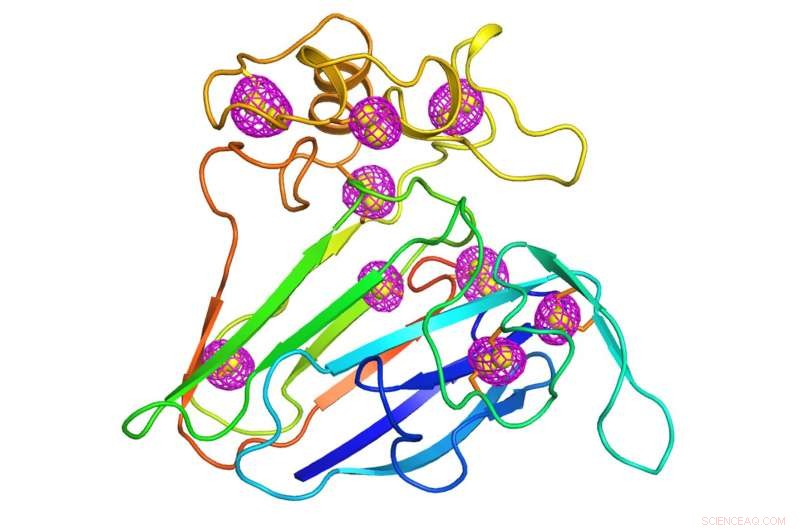
Un fumetto che rappresenta la struttura di una proteina vegetale ben studiata che è servita come banco di prova per la tecnica di microcristallografia di nuova concezione. I reticoli magenta che circondano gli atomi di zolfo intrinseci alla proteina (sfere gialle) indicano i segnali anomali che sono stati estratti utilizzando la diffrazione a raggi X a bassa energia di migliaia di cristalli che misurano meno di 10 milionesimi di metro, le dimensioni di un batterio. Credito:Brookhaven National Laboratory
L'uso dei raggi X per rivelare le strutture 3D su scala atomica delle proteine ha portato a innumerevoli progressi nella comprensione del funzionamento di queste molecole nei batteri, virus, impianti, e umani, e ha guidato lo sviluppo di farmaci di precisione per combattere malattie come il cancro e l'AIDS. Ma molte proteine non possono essere trasformate in cristalli abbastanza grandi da poter decifrare la loro disposizione atomica. Per affrontare questa sfida, gli scienziati del Brookhaven National Laboratory del Dipartimento dell'Energia degli Stati Uniti (DOE) e i colleghi della Columbia University hanno sviluppato un nuovo approccio per risolvere le strutture proteiche da minuscoli cristalli.
Il metodo si basa su un trattamento unico del campione, estrazione del segnale, e approcci di assemblaggio dei dati, e una linea di luce in grado di focalizzare intensi raggi X presso la National Synchrotron Light Source II (NSLS-II) di Brookhaven, una struttura per gli utenti dell'Office of Science del DOE, in un punto di un milionesimo di metro, circa un cinquantesimo della larghezza di un capello umano.
"La nostra tecnica apre davvero le porte alla gestione di microcristalli che prima erano inaccessibili, compresi i recettori della superficie cellulare difficili da cristallizzare e altre proteine di membrana, proteine flessibili, e molte proteine umane complesse, ", ha affermato lo scienziato del laboratorio Brookhaven Qun Liu, l'autore corrispondente dello studio, pubblicato il 3 maggio 2019, in IUCrJ , una rivista dell'Unione Internazionale di Cristallografia.
Decifrare le strutture proteiche
La cristallografia proteica è stata un metodo dominante per risolvere le strutture proteiche dal 1958, migliorando nel tempo poiché le sorgenti di raggi X sono diventate più potenti, consentendo determinazioni strutturali più precise. Per determinare una struttura proteica, gli scienziati misurano come i raggi X come quelli generati a NSLS-II diffrange, o rimbalzare, gli atomi in un reticolo cristallino ordinato costituito da molte copie della stessa molecola proteica sono tutti disposti allo stesso modo. Il modello di diffrazione trasmette informazioni su dove si trovano gli atomi. Ma non è sufficiente.
"Solo le ampiezze delle 'onde' di raggi X diffratti vengono registrate sul rivelatore, ma non le loro fasi (i tempi tra le onde), " ha detto Liu. "Entrambi sono necessari per ricostruire una struttura 3-D. Questo è il cosiddetto problema della fase cristallografica".
I cristallografi hanno risolto questo problema raccogliendo dati di fase da un diverso tipo di diffusione, noto come diffusione anomala. La dispersione anomala si verifica quando gli atomi più pesanti dei componenti principali del carbonio di una proteina, idrogeno, e l'azoto assorbono e riemettono alcuni dei raggi X. Questo accade quando l'energia dei raggi X è vicina all'energia che quegli atomi pesanti amano assorbire. Gli scienziati a volte inseriscono artificialmente atomi pesanti come il selenio o il platino nella proteina per questo scopo. Ma gli atomi di zolfo, che appaiono naturalmente in tutte le molecole proteiche, può anche produrre tali segnali, seppur più debole. Anche se questi segnali anomali sono deboli, un grande cristallo di solito ha abbastanza copie della proteina con abbastanza atomi di zolfo per renderle misurabili. Ciò fornisce agli scienziati le informazioni di fase necessarie per individuare la posizione degli atomi di zolfo e tradurre i modelli di diffrazione in una struttura tridimensionale completa.
"Una volta che conosci le posizioni dello zolfo, puoi calcolare le fasi per gli altri atomi di proteine perché il rapporto tra lo zolfo e gli altri atomi è fisso, " disse Liù.
Ma minuscoli cristalli, per definizione, non hanno così tante copie della proteina di interesse. Quindi, invece di cercare informazioni sulla diffrazione e sulla fase da copie ripetute di una proteina in un singolo grande cristallo, il team di Brookhaven/Columbia ha sviluppato un modo per effettuare misurazioni da molti minuscoli cristalli, e quindi assemblare i dati collettivi.
Piccoli cristalli, grandi risultati
Per maneggiare i minuscoli cristalli, il team ha sviluppato griglie di campioni modellate con pozzi di dimensioni micro. Dopo aver versato il solvente contenente i microcristalli su queste griglie ben montate, gli scienziati hanno rimosso il solvente e congelato i cristalli intrappolati sulle griglie.
"Abbiamo ancora una sfida, anche se, perché non possiamo vedere dove sono i minuscoli cristalli sulla nostra griglia, " disse Liu. "Per scoprirlo, abbiamo usato la microdiffrazione alla linea di luce FMX (Frontier Microfocusing Macromolecular Crystallography) di NSLS-II per esaminare l'intera griglia. Scansione riga per riga, possiamo scoprire dove sono nascosti quei cristalli."
Come Martin Fuchs, lo scienziato capo della linea di luce di FMX, spiegato, "La linea di luce FMX può focalizzare l'intera intensità del raggio di raggi X fino a una dimensione di un micron, o milionesimo di metro. Possiamo controllare finemente la dimensione del raggio per adattarla alla dimensione dei cristalli, cinque micron nel caso dell'esperimento in corso. Queste capacità sono fondamentali per ottenere il miglior segnale, " Egli ha detto.
Wuxian Shi, un altro scienziato della linea di luce FMX, ha osservato che "i dati raccolti nell'indagine sulla griglia contengono informazioni sulla posizione dei cristalli. Inoltre, possiamo anche vedere quanto bene ogni cristallo diffrange, che ci permette di scegliere solo i migliori cristalli per la raccolta dei dati."
Gli scienziati sono stati quindi in grado di manovrare il supporto del campione per posizionare ciascun microcristallo mappato di interesse al centro del raggio di raggi X di precisione per la raccolta dei dati.
Hanno usato l'energia più bassa disponibile sulla linea di luce, sintonizzata per avvicinarsi il più possibile all'energia di assorbimento degli atomi di zolfo, e hanno raccolto dati di dispersione anomala.
"La maggior parte delle linee di luce cristallografiche non è riuscita a raggiungere il limite di assorbimento dello zolfo per segnali anomali ottimizzati, ", ha detto il coautore Wayne Hendrickson della Columbia University. "Fortunatamente, NSLS-II è una sorgente di luce di sincrotrone leader a livello mondiale che fornisce raggi X luminosi che coprono un ampio spettro di energia dei raggi X. E anche se il nostro livello di energia era leggermente al di sopra dell'energia di assorbimento ideale per lo zolfo, ha generato i segnali anomali di cui avevamo bisogno."
Ma gli scienziati avevano ancora del lavoro da fare per estrarre quei segnali importanti e assemblare i dati da molti minuscoli cristalli.
"In realtà stiamo ottenendo migliaia di dati, " ha detto Liu. "Abbiamo usato circa 1400 microcristalli, ciascuno con il proprio set di dati. Dobbiamo mettere insieme tutti i dati di quei microcristalli".
They also had to weed out data from crystals that were damaged by the intense x-rays or had slight variations in atomic arrangements.
"A single microcrystal does not diffract x-rays sufficiently for structure solution prior to being damaged by the x-rays, " said Sean McSweeney, deputy photon division director and program manager of the Structural Biology Program at NSLS-II. "This is particularly true with crystals of only a few microns, the size of about a bacterial cell. We needed a way to account for that damage and crystal structure variability so it wouldn't skew our results."
They accomplished these goals with a sophisticated multi-step workflow process that sifted through the data, discarded outliers that might have been caused by radiation damage or incompatible crystals, and ultimately extracted the anomalous scattering signals.
"This is a critical step, " said Liu. "We developed a computing procedure to assure that only compatible data were merged in a way to align the individual microcrystals from diffraction patterns. That gave us the required signal-to-noise ratios for structure determination."
Applying the technique
This technique can be used to determine the structure of any protein that has proven hard to crystallize to a large size. These include cell-surface receptors that allow cells of advanced lifeforms such as animals and plants to sense and respond to the environment around them by releasing hormones, transmitting nerve signals, or secreting compounds associated with cell growth and immunity.
"To adapt to the environment through evolution, these proteins are malleable and have lots of non-uniform modifications, " said Liu. "It's hard to get a lot of repeat copies in a crystal because they don't pack well."
Negli umani, receptors are common targets for drugs, so having knowledge of their varied structures could help guide the development of new, more targeted pharmaceuticals.
But the technique is not restricted to just small crystals.
"The method we developed can handle small protein crystals, but it can also be used for any size protein crystals, any time you need to combine data from more than one sample, " ha detto Liù.