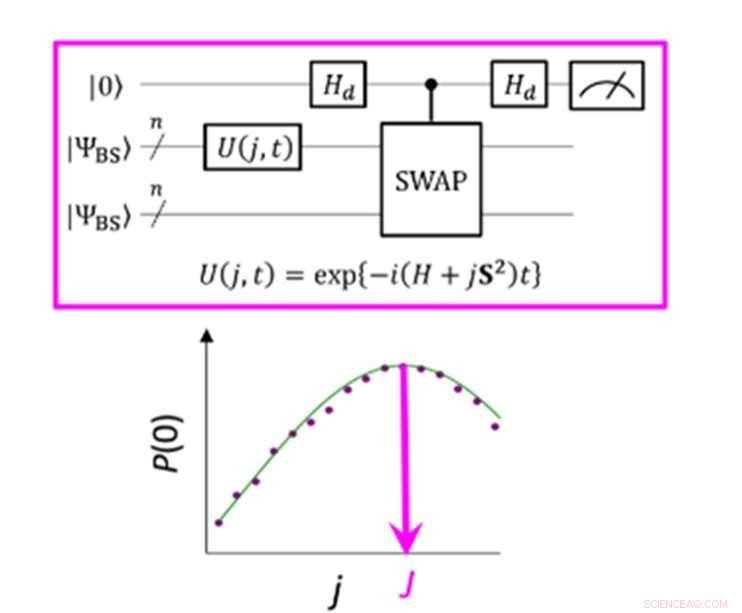
Un circuito quantistico che consente la massima probabilità di P(0) nella misurazione del parametro J. Credito:K. Sugisaki, K. Sato e T. Takui
I ricercatori dell'Università della città di Osaka utilizzano gli stati di sovrapposizione quantistica e l'inferenza bayesiana per creare un algoritmo quantistico, facilmente eseguibile su computer quantistici, che calcola in modo accurato e diretto le differenze di energia tra gli stati elettronici di base e di spin eccitato dei sistemi molecolari in tempo polinomiale.
Comprendere come funziona il mondo naturale ci consente di imitarlo a beneficio dell'umanità. Pensa a quanto ci affidiamo alle batterie. Al centro c'è la comprensione delle strutture molecolari e del comportamento degli elettroni al loro interno. Il calcolo delle differenze di energia tra la massa elettronica di una molecola e gli stati di spin eccitato ci aiuta a capire come utilizzare meglio quella molecola in una varietà di sostanze chimiche, applicazioni biomediche e industriali. Abbiamo fatto molti progressi nelle molecole con sistemi a guscio chiuso, in cui gli elettroni sono appaiati e stabili. Sistemi a guscio aperto, d'altra parte, sono meno stabili e il loro comportamento elettronico sottostante è complesso, e quindi più difficile da capire. Hanno elettroni spaiati nel loro stato fondamentale, che fanno variare la loro energia a causa della natura intrinseca degli spin elettronici, e rende difficili le misurazioni, soprattutto quando le molecole aumentano di dimensione e complessità. Sebbene tali molecole siano abbondanti in natura, mancano algoritmi in grado di gestire questa complessità. Un ostacolo è stato affrontare quella che viene chiamata l'esplosione esponenziale del tempo computazionale. Usare un computer convenzionale per calcolare come gli spin spaiati influenzano l'energia di una molecola a guscio aperto richiederebbero centinaia di milioni di anni, tempo che gli umani non hanno.
I computer quantistici sono in fase di sviluppo per aiutare a ridurre questo a quello che viene chiamato "tempo polinomiale". Però, il processo utilizzato dagli scienziati per calcolare le differenze di energia delle molecole a guscio aperto è stato essenzialmente lo stesso sia per i computer convenzionali che per quelli quantistici. Ciò ostacola l'uso pratico dell'informatica quantistica nelle applicazioni chimiche e industriali.
"Gli approcci che invocano i veri algoritmi quantistici ci aiutano a trattare i sistemi a guscio aperto in modo molto più efficiente rispetto all'utilizzo di computer classici, " dichiarano Kenji Sugisaki e Takeji Takui della Osaka City University. Con i loro colleghi, hanno sviluppato un algoritmo quantistico eseguibile su computer quantistici, quale può, per la prima volta, calcolare accuratamente le differenze di energia tra gli stati elettronici di massa e di spin eccitato dei sistemi molecolari a guscio aperto. I loro risultati sono stati pubblicati sulla rivista Scienze chimiche il 24 dicembre 2020.
La differenza di energia tra gli stati di spin molecolare è caratterizzata dal valore del parametro di interazione di scambio J. Gli algoritmi quantistici convenzionali sono stati in grado di calcolare accuratamente le energie per le molecole a guscio chiuso "ma non sono stati in grado di gestire sistemi con una forte multiconfigurazione carattere, " afferma il gruppo. Fino ad ora, gli scienziati hanno ipotizzato che per ottenere il parametro J si debba prima calcolare l'energia totale di ogni stato di spin. Nelle molecole a guscio aperto questo è difficile perché l'energia totale di ogni stato di spin varia notevolmente al variare dell'attività e delle dimensioni della molecola. Però, "la differenza di energia in sé non è molto dipendente dalle dimensioni del sistema, " fa notare il team di ricerca. Ciò li ha portati a creare un algoritmo con calcoli che si concentravano sulla differenza di spin, non i singoli stati di spin. La creazione di un tale algoritmo richiedeva di lasciar andare i presupposti sviluppati da anni di utilizzo di computer convenzionali e concentrarsi sulle caratteristiche uniche del calcolo quantistico, vale a dire "stati di sovrapposizione quantistica".
La "sovrapposizione" consente agli algoritmi di rappresentare due variabili contemporaneamente, che quindi consente agli scienziati di concentrarsi sulla relazione tra queste variabili senza la necessità di determinare prima i loro stati individuali. Il team di ricerca ha utilizzato una funzione d'onda a simmetria spezzata come sovrapposizione di funzioni d'onda con diversi stati di spin e l'ha riscritta nell'equazione hamiltoniana per il parametro J. Eseguendo questo nuovo circuito quantistico, il team è stato in grado di concentrarsi sulle deviazioni dal proprio obiettivo e applicando l'inferenza bayesiana, una tecnica di apprendimento automatico, hanno portato queste deviazioni per determinare il parametro di interazione di scambio J. "Sono state eseguite simulazioni numeriche basate su questo metodo per la dissociazione covalente dell'idrogeno molecolare (H 2 ), la dissociazione del triplo legame dell'azoto molecolare (N 2 ), e gli stati fondamentali di C, Oh, atomi di Si e NH, OH + , CH 2 , NF e O 2 molecole con un errore inferiore a 1 kcal/mol, " aggiunge il gruppo di ricerca.
"Abbiamo in programma di installare il nostro calcolatore dei parametri di accoppiamento eXchange bayesiano con il software per funzioni d'onda a simmetria spezzata (BxB) su computer quantistici a breve termine dotati di dispositivi quantistici rumorosi (nessuna correzione degli errori quantistici) su scala intermedia (diverse centinaia di qubit) (dispositivi NISQ). ), testare l'utilità per i calcoli della chimica quantistica di sistemi molecolari di dimensioni reali".