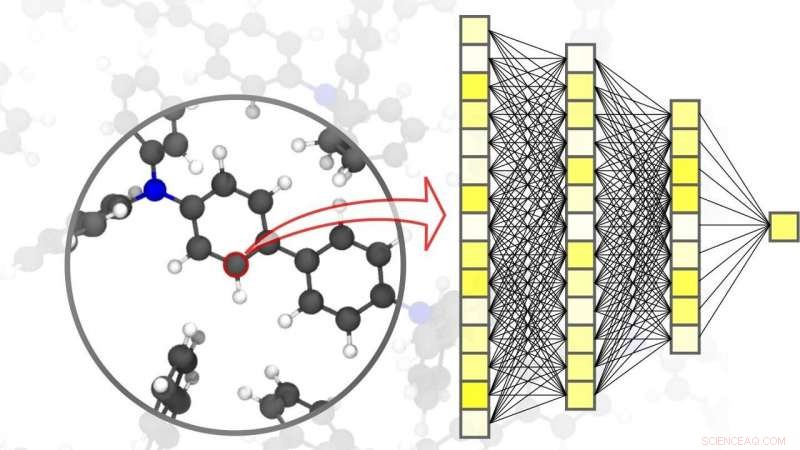
Le reti neurali consentono simulazioni precise nella scienza dei materiali, fino al livello dei singoli atomi. Credito:Pascal Friederich, KIT
Ricerca, sviluppo, e la produzione di nuovi materiali dipendono fortemente dalla disponibilità di metodi di simulazione rapidi e allo stesso tempo accurati. Apprendimento automatico, in cui l'intelligenza artificiale (AI) acquisisce e applica autonomamente nuove conoscenze, consentirà presto ai ricercatori di sviluppare sistemi di materiali complessi in un ambiente puramente virtuale. Come funziona, e quali applicazioni ne beneficeranno? In un articolo pubblicato su Materiali della natura rivista, spiegano tutto un ricercatore del Karlsruhe Institute of Technology (KIT) e i suoi colleghi di Göttingen e Toronto.
La digitalizzazione e la virtualizzazione stanno diventando sempre più importanti in un'ampia gamma di discipline scientifiche. Una di queste discipline è la scienza dei materiali:ricerca, sviluppo, e la produzione di nuovi materiali dipendono fortemente dalla disponibilità di metodi di simulazione rapidi e allo stesso tempo accurati. Questo, a sua volta, è vantaggioso per un'ampia gamma di applicazioni diverse, dai sistemi di accumulo di energia efficienti, come quelle indispensabili per l'utilizzo delle energie rinnovabili, a nuovi farmaci, per il cui sviluppo è necessaria la comprensione di processi biologici complessi. L'intelligenza artificiale e i metodi di apprendimento automatico possono portare le simulazioni nelle scienze dei materiali a un livello superiore. "Rispetto ai metodi di simulazione convenzionali basati su calcoli meccanici classici o quantistici, l'uso di reti neurali specificamente adattate alle simulazioni di materiali ci consente di ottenere un significativo vantaggio in termini di velocità, " spiega il fisico ed esperto di intelligenza artificiale, il professor Pascal Friederich, Capo del gruppo di ricerca AiMat—Artificial Intelligence for Materials Sciences presso l'Institute of Theoretical Informatics (ITI) del KIT. "Con sistemi di simulazione più veloci, gli scienziati saranno in grado di sviluppare sistemi materiali più grandi e complessi in un ambiente puramente virtuale, e per comprenderli e ottimizzarli fino al livello atomico."
Alta precisione dall'atomo al materiale
In un articolo pubblicato su Materiali della natura , Pascal Friederich, che è anche capogruppo associato della divisione Nanomaterials by Information-Guided Design presso l'Institute of Nanotechnology (INT) di KIT, regali, insieme a ricercatori dell'Università di Göttingen e dell'Università di Toronto, una panoramica dei principi di base dell'apprendimento automatico utilizzati per le simulazioni nelle scienze dei materiali. Ciò include anche il processo di acquisizione dei dati e i metodi di apprendimento attivo. Gli algoritmi di apprendimento automatico non solo consentono all'intelligenza artificiale di elaborare i dati di input, ma anche per trovare modelli e correlazioni in grandi insiemi di dati, imparare da loro, e fare previsioni e decisioni autonome. Per le simulazioni nella scienza dei materiali, è importante ottenere un'elevata precisione su diverse scale temporali e dimensionali, che vanno dall'atomo alla materia, limitando i costi di calcolo. Nel loro articolo, gli scienziati discutono anche di varie applicazioni attuali, come piccole molecole organiche e grandi biomolecole, solido strutturalmente disordinato, liquido, e materiali gassosi, così come sistemi cristallini complessi, ad esempio, strutture metallo-organiche che possono essere utilizzate per lo stoccaggio del gas o per la separazione, per sensori o per catalizzatori.
Ancora più velocità con i metodi ibridi
Per ampliare ulteriormente le possibilità di simulazioni dei materiali in futuro, i ricercatori di Karlsruhe, Gottinga, e Toronto suggeriscono lo sviluppo di metodi ibridi:questi combinano metodi di machine learning (ML) e di meccanica molecolare (MM). Le simulazioni MM utilizzano i cosiddetti campi di forza per calcolare le forze che agiscono su ogni singola particella e quindi prevedere i movimenti. Poiché le potenzialità dei metodi ML e MM sono abbastanza simili, è possibile una stretta integrazione con aree di transizione variabili. Questi metodi ibridi potrebbero accelerare significativamente la simulazione di grandi biomolecole o reazioni enzimatiche in futuro, Per esempio.