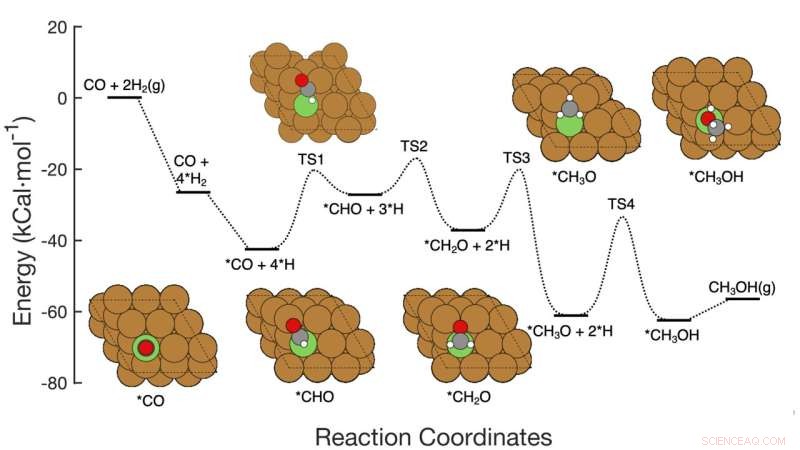
Questo grafico mostra il percorso di reazione in sette fasi dell'idrogenazione della CO al metanolo su catalizzatori a base di rame, inclusi i reagenti in ogni fase, le disposizioni atomiche schematiche degli intermedi e le barriere di attivazione dell'energia necessarie per passare da una fase all'altra. Il team di Brookhaven Lab ha dimostrato un framework di apprendimento automatico che ha identificato con successo quali passaggi/combinazioni di passaggi modificare per migliorare la produzione di metanolo. Il loro lavoro potrebbe aiutare a guidare la progettazione di nuovi catalizzatori per raggiungere tale obiettivo e la struttura può essere applicata per ottimizzare altre reazioni. Credito:Brookhaven National Laboratory
I chimici del Brookhaven National Laboratory del Dipartimento dell'Energia degli Stati Uniti hanno sviluppato un nuovo framework di apprendimento automatico (ML) in grado di individuare le fasi di una conversione chimica multifase da modificare per migliorare la produttività. L'approccio potrebbe aiutare a guidare la progettazione di catalizzatori, "distributori" chimici che accelerano le reazioni.
Il team ha sviluppato il metodo per analizzare la conversione del monossido di carbonio (CO) in metanolo utilizzando un catalizzatore a base di rame. La reazione consiste in sette passaggi elementari abbastanza semplici.
"Il nostro obiettivo era identificare quale passaggio elementare nella rete di reazione o quale sottoinsieme di passaggi controlla l'attività catalitica", ha affermato Wenjie Liao, il primo autore di un articolo che descrive il metodo appena pubblicato sulla rivista Catalysis Science &Technology . Liao è uno studente laureato presso la Stony Brook University che ha lavorato con gli scienziati nel gruppo Catalysis Reactivity and Structure (CRS) nella divisione di chimica del Brookhaven Lab.
Ping Liu, il chimico del CRS che ha guidato il lavoro, ha dichiarato:"Abbiamo usato questa reazione come esempio del nostro metodo framework ML, ma puoi inserire qualsiasi reazione in questo framework in generale".
Targeting delle energie di attivazione
Immagina una reazione chimica a più fasi come montagne russe con colline di diverse altezze. L'altezza di ogni collina rappresenta l'energia necessaria per passare da un gradino all'altro. I catalizzatori abbassano queste "barriere di attivazione" facilitando l'unione dei reagenti o consentendo loro di farlo a temperature o pressioni inferiori. Per accelerare la reazione complessiva, un catalizzatore deve mirare alla fase o alle fasi che hanno l'impatto maggiore.
Tradizionalmente, gli scienziati che cercano di migliorare tale reazione calcolano come cambiare ciascuna barriera di attivazione una alla volta potrebbe influenzare il tasso di produzione complessivo. Questo tipo di analisi potrebbe identificare quale fase era "limitante la velocità" e quali fasi determinano la selettività della reazione, ovvero se i reagenti procedono al prodotto desiderato o lungo un percorso alternativo verso un sottoprodotto indesiderato.

Il chimico del Brookhaven Lab Ping Liu e Wenjie Liao, uno studente laureato alla Stony Brook University, hanno sviluppato un framework di apprendimento automatico per identificare quali fasi della reazione chimica potrebbero essere mirate per migliorare la produttività della reazione. Credito:Brookhaven National Laboratory
Ma, secondo Liu, "Queste stime finiscono per essere molto approssimative con molti errori per alcuni gruppi di catalizzatori. Ciò ha davvero danneggiato la progettazione e lo screening dei catalizzatori, che è ciò che stiamo cercando di fare", ha affermato.
Il nuovo framework di apprendimento automatico è progettato per migliorare queste stime in modo che gli scienziati possano prevedere meglio come i catalizzatori influenzeranno i meccanismi di reazione e la produzione chimica.
"Ora, invece di spostare una barriera alla volta, stiamo spostando tutte le barriere contemporaneamente. E utilizziamo l'apprendimento automatico per interpretare quel set di dati", ha affermato Liao.
Questo approccio, ha affermato il team, fornisce risultati molto più affidabili, incluso il modo in cui le fasi di una reazione interagiscono.
"In condizioni di reazione, questi passaggi non sono isolati o separati l'uno dall'altro; sono tutti collegati", ha affermato Liu. "Se esegui solo un passaggio alla volta, perdi molte informazioni:le interazioni tra i passaggi elementari. Questo è ciò che è stato catturato in questo sviluppo", ha affermato.
Costruire il modello
Gli scienziati hanno iniziato costruendo un set di dati per addestrare il loro modello di apprendimento automatico. Il set di dati si basava sui calcoli della "teoria del funzionale della densità" (DFT) dell'energia di attivazione richiesta per trasformare una disposizione di atomi nella successiva attraverso i sette passaggi della reazione. Quindi gli scienziati hanno eseguito simulazioni al computer per esplorare cosa sarebbe successo se avessero cambiato tutte e sette le barriere di attivazione contemporaneamente, alcune salendo, altre scendendo, alcune individualmente e altre in coppia.
"La gamma di dati che abbiamo incluso si basava sull'esperienza precedente con queste reazioni e questo sistema catalitico, all'interno dell'interessante gamma di variazione che probabilmente ti darà prestazioni migliori", ha affermato Liu.
Simulando le variazioni in 28 "descrittori", comprese le energie di attivazione per i sette passaggi più coppie di passaggi che cambiano due alla volta, il team ha prodotto un set di dati completo di 500 punti dati. Questo set di dati prevedeva come tutte quelle modifiche individuali e coppie di modifiche avrebbero influenzato la produzione di metanolo. Il modello ha quindi valutato i 28 descrittori in base alla loro importanza nel guidare la produzione di metanolo.
"Il nostro modello ha 'imparato' dai dati e ha identificato sei descrittori chiave che prevede avrebbero il maggiore impatto sulla produzione", ha affermato Liao.
Dopo che i descrittori importanti sono stati identificati, gli scienziati hanno riqualificato il modello ML utilizzando solo quei sei descrittori "attivi". Questo modello ML migliorato è stato in grado di prevedere l'attività catalitica basata esclusivamente sui calcoli DFT per quei sei parametri.
"Invece di dover calcolare tutti i 28 descrittori, ora puoi calcolare solo con i sei descrittori e ottenere i tassi di conversione del metanolo che ti interessano", ha affermato Liu.
Il team afferma che possono anche utilizzare il modello per selezionare i catalizzatori. Se riescono a progettare un catalizzatore che migliora il valore dei sei descrittori attivi, il modello prevede un tasso massimo di produzione di metanolo.
Comprendere i meccanismi
Quando il team ha confrontato le previsioni del proprio modello con le prestazioni sperimentali del catalizzatore e le prestazioni di leghe di vari metalli con il rame, le previsioni si sono abbinate ai risultati sperimentali. I confronti dell'approccio ML con il metodo precedente utilizzato per prevedere le prestazioni delle leghe hanno mostrato che il metodo ML è di gran lunga superiore.
I dati hanno anche rivelato molti dettagli su come i cambiamenti nelle barriere energetiche potrebbero influenzare il meccanismo di reazione. Di particolare interesse e importanza è stato il modo in cui le diverse fasi della reazione lavorano insieme. Ad esempio, i dati hanno mostrato che in alcuni casi, l'abbassamento della barriera energetica nella sola fase di limitazione della velocità non migliorerebbe di per sé la produzione di metanolo. Ma modificando la barriera energetica di una fase precedente nella rete di reazione, mantenendo l'energia di attivazione della fase di limitazione della velocità entro un intervallo ideale, aumenterebbe la produzione di metanolo.
"Il nostro metodo ci fornisce informazioni dettagliate che potremmo essere in grado di utilizzare per progettare un catalizzatore che coordini bene l'interazione tra questi due passaggi", ha affermato Liu.
Ma Liu è molto entusiasta del potenziale per l'applicazione di tali framework ML basati sui dati a reazioni più complicate.
"Abbiamo usato la reazione del metanolo per dimostrare il nostro metodo. Ma il modo in cui genera il database e il modo in cui addestriamo il modello ML e come interpoliamo il ruolo della funzione di ciascun descrittore per determinare il peso complessivo in termini di importanza, questo può essere applicato facilmente ad altre reazioni", ha detto. + Esplora ulteriormente