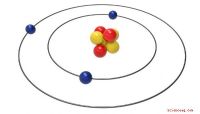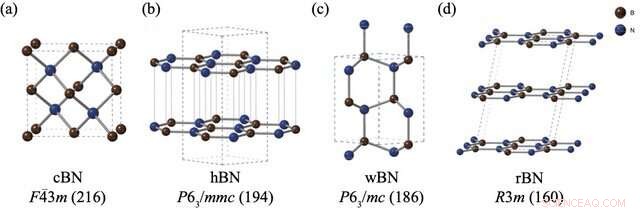
Le strutture e i gruppi spaziali di (a) nitruro di boro miscela di zinco (cBN), (b) nitruro di boro esagonale (hBN), (c) nitruro di boro wurtzite (wBN) e (d) nitruro di boro romboedrico (rBN). Gli atomi di boro e azoto sono raffigurati rispettivamente in marrone e blu. Credito:Kousuke Nakano di JAIST.
Il nitruro di boro (BN) è un materiale versatile con applicazioni in una varietà di campi ingegneristici e scientifici. Ciò è in gran parte dovuto a un'interessante proprietà del BN chiamata "polimorfismo", caratterizzata dalla capacità di cristallizzare in più di un tipo di struttura. Ciò si verifica generalmente in risposta a variazioni di temperatura, pressione o entrambe. Inoltre, le diverse strutture, chiamate "polimorfi", differiscono notevolmente nelle loro proprietà fisiche pur avendo la stessa formula chimica. Di conseguenza, i polimorfi svolgono un ruolo importante nella progettazione dei materiali e la conoscenza di come favorire selettivamente la formazione del polimorfo desiderato è fondamentale a questo proposito.
Tuttavia, i polimorfi BN pongono un problema particolare. Nonostante abbia condotto diversi esperimenti per valutare la stabilità relativa dei polimorfi BN, non è emerso un consenso su questo argomento. Sebbene i metodi computazionali siano spesso l'approccio di riferimento per questi problemi, i polimorfi BN hanno posto serie sfide alle tecniche di calcolo standard a causa delle deboli "interazioni di van der Waals (vdW)" tra i loro livelli, che non sono considerate in questi calcoli. Inoltre, i quattro polimorfi BN stabili, vale a dire romboedrico (rBN), esagonale (hBN), wurtzite (wBN) e zinco-blenda (cBN), si manifestano all'interno di un ristretto intervallo di energia, rendendo la cattura di piccole differenze di energia insieme alle interazioni vdW ancora più impegnativo.
Un team di ricerca internazionale guidato dall'assistente professore Kousuke Nakano del Japan Advanced Institute of Science and Technology (JAIST) ha ora fornito prove per risolvere il dibattito. Nel loro studio, hanno affrontato il problema con un framework di calcolo dei primi principi all'avanguardia, vale a dire simulazioni Monte Carlo a diffusione di nodi fissi (FNDMC). FNDMC rappresenta un passaggio nel popolare metodo di simulazione quantistica Monte Carlo, in cui una "funzione d'onda" quantistica parametrizzata a molti corpi viene prima ottimizzata per raggiungere lo stato fondamentale e quindi fornita all'FNDMC.
Inoltre, il team ha anche calcolato l'energia di Gibbs (il lavoro utile ottenibile da un sistema a pressione e temperatura costanti) dei polimorfi BN per diverse temperature e pressioni utilizzando la teoria del funzionale della densità (DFT) e calcoli fononici. Questo documento è stato reso disponibile online il 24 marzo 2022 e pubblicato su The Journal of Physical Chemistry C .
Secondo i risultati di FNDMC, hBN era la struttura più stabile, seguita da rBN, cBN e wBN. Questi risultati erano coerenti sia a 0 K che a 300 K (temperatura ambiente). Tuttavia, le stime DFT hanno prodotto risultati contrastanti per due diverse approssimazioni. Il Dr. Nakano spiega questi risultati contraddittori:"I nostri risultati rivelano che la stima delle stabilità relative è fortemente influenzata dal funzionale correlazionale di scambio o dall'approssimazione utilizzata nel calcolo DFT. Di conseguenza, non è possibile raggiungere una conclusione quantitativa utilizzando i risultati DFT, ed è necessario un approccio più accurato, come FNDMC."
In particolare, i risultati di FNDMC erano in accordo con quelli generati da altri metodi di calcolo raffinati, come "cluster accoppiato", suggerendo che FNDMC è uno strumento efficace per trattare i polimorfi, in particolare quelli governati dalle forze vdW. Il team ha anche dimostrato che può fornire altre informazioni importanti, come energie di riferimento affidabili, quando i dati sperimentali non sono disponibili.
Il Dr. Nakano è entusiasta delle prospettive future del metodo nell'area della scienza dei materiali. "Il nostro studio dimostra la capacità di FNDMC di rilevare minuscole variazioni di energia che coinvolgono le forze vdW, che stimoleranno l'uso di questo metodo per altri materiali di van der Waals", afferma. "Inoltre, le simulazioni molecolari basate su questo metodo accurato e affidabile potrebbero potenziare la progettazione dei materiali, consentendo lo sviluppo di farmaci e catalizzatori". + Esplora ulteriormente