I ricercatori della Carnegie Mellon University e del Los Alamos National Laboratory hanno utilizzato l'apprendimento automatico per creare un modello in grado di simulare processi reattivi in una serie diversificata di materiali e condizioni organici.
"È uno strumento che può essere utilizzato per indagare su più reazioni in questo campo", ha affermato Shuhao Zhang, uno studente laureato presso il Dipartimento di Chimica della Carnegie Mellon University. "Siamo in grado di offrire una simulazione completa dei meccanismi di reazione."
Zhang è il primo autore dell'articolo che spiega la creazione e i risultati di questo nuovo modello di machine learning intitolato "Exploring the Frontiers of Chemistry with a General Reactive Machine Learning Potential", pubblicato su Nature Chemistry .
Sebbene i ricercatori abbiano già simulato reazioni, i metodi precedenti presentavano molteplici problemi. I modelli di campi di forza reattivi sono relativamente comuni, ma di solito richiedono una formazione per tipi di reazione specifici. I modelli tradizionali che utilizzano la meccanica quantistica, in cui le reazioni chimiche vengono simulate in base alla fisica sottostante, possono essere applicati a qualsiasi materiale e molecola, ma questi modelli richiedono l'utilizzo di supercomputer.
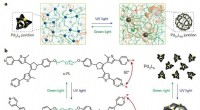
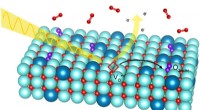
Questo nuovo potenziale interatomico di apprendimento automatico generale (ANI-1xnr) può eseguire simulazioni per materiali arbitrari contenenti gli elementi carbonio, idrogeno, azoto e ossigeno e richiede potenza di calcolo e tempo significativamente inferiori rispetto ai tradizionali modelli di meccanica quantistica. Secondo Olexandr Isayev, professore associato di chimica alla Carnegie Mellon e capo del laboratorio in cui è stato sviluppato il modello, questa svolta è dovuta agli sviluppi nell'apprendimento automatico.
"L'apprendimento automatico sta emergendo come un potente approccio per costruire varie forme di potenziali atomistici trasferibili utilizzando algoritmi di regressione. L'obiettivo generale di questo progetto è sviluppare un metodo di apprendimento automatico in grado di prevedere l'energia e le velocità di reazione per i processi chimici con elevata precisione, ma con un costo computazionale molto basso," ha detto Isayev.
"Abbiamo dimostrato che questi modelli di apprendimento automatico possono essere addestrati ad alti livelli di teoria della meccanica quantistica e possono prevedere con successo energie e forze con precisione meccanica quantistica e un aumento della velocità fino a 6-7 ordini di grandezza. Questo è un nuovo paradigma nelle simulazioni reattive."
I ricercatori hanno testato ANI-1xnr su diversi problemi chimici, tra cui il confronto degli additivi per biocarburanti e il monitoraggio della combustione del metano. Hanno persino ricreato l'esperimento Miller, un famoso esperimento chimico inteso a dimostrare come ha avuto origine la vita sulla Terra. Usando questo esperimento, hanno scoperto che il modello ANI-1xnr produceva risultati accurati nei sistemi a fase condensata.
Zhang ha affermato che il modello potrebbe essere potenzialmente utilizzato per altri settori della chimica con ulteriore formazione.
"Abbiamo scoperto che può essere potenzialmente utilizzato per simulare processi biochimici come le reazioni enzimatiche", ha detto Zhang. "Non l'abbiamo progettato per essere utilizzato in questo modo, ma dopo la modifica potrebbe essere utilizzato per quello scopo."
In futuro, il team prevede di perfezionare ANI-1xnr e consentirgli di funzionare con più elementi e in più aree chimiche, e cercherà di aumentare la scala delle reazioni che può elaborare. Ciò potrebbe consentirne l'utilizzo in molteplici campi in cui la progettazione di nuove reazioni chimiche potrebbe essere rilevante, come la scoperta di farmaci.
A Zhang e Isayev si sono uniti in questo studio Małgorzata Z. Makoś, Ryan B. Jadrich, Elfi Kraka, Kipton Barros, Benjamin T. Nebgen, Sergei Tretiak, Nicholas Lubbers, Richard A. Messerly e Justin S. Smith.
Ulteriori informazioni: Esplorare le frontiere della chimica con un potenziale generale di apprendimento automatico reattivo, Chimica naturale (2024). DOI:10.1038/s41557-023-01427-3
Informazioni sul giornale: Chimica della Natura
Fornito dalla Carnegie Mellon University