I ricercatori hanno sviluppato una piattaforma che combina esperimenti automatizzati con l'intelligenza artificiale per prevedere come le sostanze chimiche reagiranno tra loro, il che potrebbe accelerare il processo di progettazione di nuovi farmaci.
Prevedere come reagiranno le molecole è vitale per la scoperta e la produzione di nuovi farmaci, ma storicamente questo è stato un processo basato su tentativi ed errori e le reazioni spesso falliscono. Per prevedere come reagiranno le molecole, i chimici solitamente simulano elettroni e atomi in modelli semplificati, un processo computazionalmente costoso e spesso impreciso.
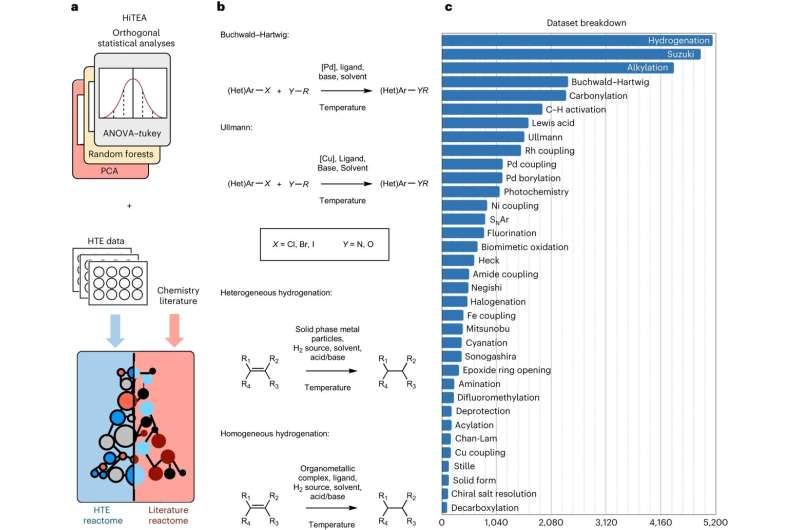
Ora, i ricercatori dell’Università di Cambridge hanno sviluppato un approccio basato sui dati, ispirato alla genomica, in cui gli esperimenti automatizzati vengono combinati con l’apprendimento automatico per comprendere la reattività chimica, accelerando notevolmente il processo. Hanno chiamato il loro approccio, che è stato convalidato su un set di dati di oltre 39.000 reazioni rilevanti dal punto di vista farmaceutico, "reattoma" chimico.
I loro risultati, riportati sulla rivista Nature Chemistry , sono il prodotto di una collaborazione tra Cambridge e Pfizer.
"Il reattore potrebbe cambiare il modo in cui pensiamo alla chimica organica", ha affermato la dott.ssa Emma King-Smith del Cavendish Laboratory di Cambridge, la prima autrice dell'articolo. "Una comprensione più approfondita della chimica potrebbe consentirci di realizzare prodotti farmaceutici e tanti altri prodotti utili molto più velocemente. Ma, cosa più importante, la comprensione che speriamo di generare sarà vantaggiosa per chiunque lavori con le molecole."
L'approccio del reattoma individua le correlazioni rilevanti tra reagenti, reagenti e prestazioni della reazione dai dati e sottolinea le lacune nei dati stessi. I dati vengono generati da esperimenti automatizzati molto veloci o ad alto rendimento.
"La chimica ad alta produttività ha rappresentato un punto di svolta, ma credevamo che ci fosse un modo per scoprire una comprensione più profonda delle reazioni chimiche rispetto a quella che può essere osservata dai risultati iniziali di un esperimento ad alta produttività", ha affermato King-Smith.
"Il nostro approccio svela le relazioni nascoste tra i componenti della reazione e i risultati", ha affermato il dottor Alpha Lee, che ha guidato la ricerca. "Il set di dati su cui abbiamo addestrato il modello è enorme:contribuirà a portare il processo di scoperta chimica da tentativi ed errori all'era dei big data."
In un articolo correlato, pubblicato su Nature Communications , il team ha sviluppato un approccio di apprendimento automatico che consente ai chimici di introdurre trasformazioni precise in regioni molecolari pre-specificate, consentendo una progettazione dei farmaci più rapida.
L'approccio consente ai chimici di modificare molecole complesse, come una modifica di progettazione dell'ultimo minuto, senza doverle creare da zero. Creare una molecola in laboratorio è tipicamente un processo in più fasi, come costruire una casa. Se i chimici vogliono variare il nucleo di una molecola, il modo convenzionale è ricostruire la molecola, come abbattere una casa e ricostruirla da zero. Tuttavia, le variazioni fondamentali sono importanti per la progettazione dei farmaci.
Una classe di reazioni note come reazioni di funzionalizzazione in fase avanzata tenta di introdurre direttamente trasformazioni chimiche nel nucleo, evitando la necessità di ricominciare da zero. Tuttavia, è difficile rendere selettiva e controllata la funzionalizzazione in fase avanzata:in genere ci sono molte regioni delle molecole che possono reagire ed è difficile prevederne il risultato.
"Le funzionalizzazioni in fase avanzata possono produrre risultati imprevedibili e gli attuali metodi di modellazione, inclusa la nostra intuizione esperta, non sono perfetti", ha affermato King-Smith. "Un modello più predittivo ci darebbe l'opportunità di effettuare uno screening migliore."
I ricercatori hanno sviluppato un modello di apprendimento automatico che prevede dove una molecola reagirebbe e come il sito di reazione varierebbe in funzione delle diverse condizioni di reazione. Ciò consente ai chimici di trovare modi per modificare con precisione il nucleo di una molecola.
"Abbiamo pre-addestrato il modello su un ampio insieme di dati spettroscopici, insegnando efficacemente la chimica generale del modello, prima di perfezionarlo per prevedere queste complesse trasformazioni", ha affermato King-Smith. Questo approccio ha permesso al team di superare il limite dei pochi dati disponibili:nella letteratura scientifica sono riportate relativamente poche reazioni di funzionalizzazione in fase avanzata. Il team ha convalidato sperimentalmente il modello su una serie diversificata di molecole simili a farmaci ed è stato in grado di prevedere con precisione i siti di reattività in diverse condizioni.
"L'applicazione dell'apprendimento automatico alla chimica è spesso ostacolata dal problema che la quantità di dati è piccola rispetto alla vastità dello spazio chimico", ha affermato Lee. "Il nostro approccio, ovvero la progettazione di modelli che apprendono da set di dati di grandi dimensioni simili ma non uguali al problema che stiamo cercando di risolvere, risolve questa sfida fondamentale con pochi dati e potrebbe sbloccare progressi oltre la funzionalizzazione in fase avanzata."
Ulteriori informazioni: Emma King-Smith et al, Sondaggio del "reattoma" chimico con dati di sperimentazione ad alto rendimento, Nature Chemistry (2024). DOI:10.1038/s41557-023-01393-w
Funzionalizzazione predittiva dei Minisci in fase avanzata con apprendimento di trasferimento, Comunicazioni con la natura (2024). DOI:10.1038/s41467-023-42145-1. www.nature.com/articles/s41467-023-42145-1
Informazioni sul giornale: Comunicazioni sulla natura , Chimica della Natura
Fornito dall'Università di Cambridge