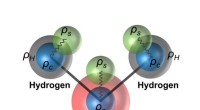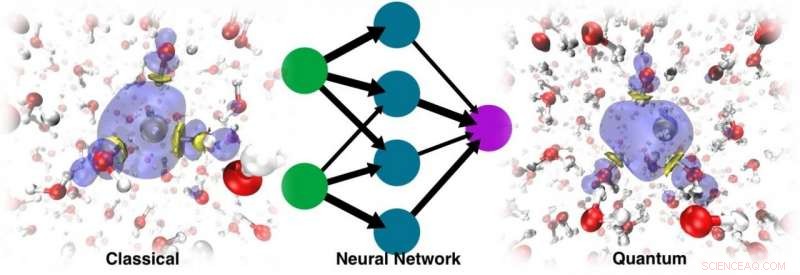
La dinamica eseguita con il risultante ML PES non solo è stata in grado di recuperare la cavità stabile, ma potrebbe anche tracciare le corrette dinamiche di localizzazione Credit:@Vladimir Rybkin
Il comportamento dell'elettrone solvatato e-aq ha implicazioni fondamentali per l'elettrochimica, fotochimica, chimica ad alta energia, così come per la biologia - il suo precursore di non equilibrio è responsabile del danno da radiazioni al DNA - ed è stato comprensibilmente oggetto di indagine sperimentale e teorica per più di 50 anni.
Sebbene l'elettrone idrato sembri semplice - è l'anione più piccolo possibile e anche l'agente riducente più semplice in chimica - catturare la sua fisica è... difficile. Sono di breve durata e generati in piccole quantità e quindi impossibili da concentrare e isolare. La loro struttura è quindi impossibile da catturare con l'osservazione sperimentale diretta come i metodi di diffrazione o NMR. La modellazione teorica si è rivelata altrettanto impegnativa.
La teoria del funzionale della densità (DFT) è il metodo della struttura elettronica più spesso utilizzato per studiare l'elettrone solvatato e l'acqua. Tuttavia, i funzionali di densità standard soffrono di errori di delocalizzazione, rendendo impossibile modellare i radicali con precisione. L'acqua pura complica notevolmente le approssimazioni DFT, sebbene la scelta dei funzionali giusti possa portare a risultati accettabili rispetto a benchmark e valori di strutture elettroniche di alto livello osservabili sperimentalmente. Una descrizione accurata dell'acqua liquida può essere ottenuta anche con metodi di chimica quantistica a molti corpi, ma sono estremamente costosi.
Sebbene una recente scoperta basata sulla dinamica molecolare su scala di picosecondi senza precedenti in termini di complessità e che richieda risorse computazionali ai limiti di ciò che è possibile ha fornito un argomento cruciale a favore di una struttura a cavità per e-aq, non ha prodotto altre nuove intuizioni o una descrizione statistica completa. La caratterizzazione completa delle proprietà del sistema richiede tempi molto più lunghi, ma la simulazione dei nuclei quantistici a questo livello della teoria della struttura elettronica è attualmente al di fuori della portata computazionale.
Il modo moderno di aggirare questo problema prevede l'uso dell'apprendimento automatico. L'addestramento di un campo di forza ML o di una superficie di energia potenziale (PES) basato su dati ab initio consente simulazioni MD molto più lunghe perché il costo di valutazione di tali energie e forze è quasi trascurabile rispetto a quello associato ai calcoli della struttura elettronica. Il problema è che l'elettrone solvatato è una specie atipica. Non ha una formula atomistica, il che pone un problema perché i PES di apprendimento automatico funzionano con rappresentazioni atomistiche.
Nel documento "Simulating the Ghost:Quantum Dynamics of the Solvated Electron, " Il ricercatore dell'Università di Zurigo Vladimir Rybkin, il dottorando Jinggang Lan e la docente Marcella Iannuzzi hanno unito la loro esperienza nella struttura elettronica e negli elettroni solvatati con la conoscenza del professore dell'EPFL Michele Ceriotti e del suo ex dottorato di ricerca. studenti Venkat Kapil, ora ricercatore presso l'Università di Cambridge, e Piero Gasparotto, ora ricercatore all'Empa, nell'apprendimento automatico e nella dinamica quantistica. Quella, con il contributo di altri colleghi, ha portato all'applicazione dell'approccio ML ai dati acquisiti da un metodo di chimica quantistica a molti corpi noto come teoria delle perturbazioni di Møller-Plesset di secondo ordine (MP2), un metodo che dia una descrizione accurata dell'acqua, comunque, senza alcun trattamento speciale dell'elettrone in eccesso.
Sono rimasti sorpresi nello scoprire che il modello era in grado di apprendere la presenza dell'elettrone solvatato come un fattore che distorceva la struttura dell'acqua liquida pura. La dinamica eseguita con il risultante ML PES non solo è stata in grado di recuperare la cavità stabile, ma potrebbe anche tracciare le corrette dinamiche di localizzazione, a partire dall'eccesso di elettrone delocalizzato aggiunto all'acqua. Alla fine, ML ha simulato l'elettrone come una sorta di "particella fantasma" che non era esplicitamente presente nel modello.
Ciò ha permesso ai ricercatori di ottenere una scala temporale di diverse centinaia di picosecondi e di raccogliere statistiche affidabili eseguendo molte traiettorie classiche computazionalmente economiche e calcolando spettri vibrazionali, strutture e diffusione. L'approccio ML ha anche permesso loro di simulare i nuclei quantistici piuttosto che classici con dinamica molecolare path-integral (PIMD). Questa tecnica è almeno un ordine di grandezza computazionalmente più costosa della MD classica e non può essere eseguita senza ML PES ad un alto livello di teoria della struttura elettronica.
Tenendo conto degli effetti quantistici nucleari, abbiamo fornito spettri vibrazionali accurati, consentendo ai ricercatori di quantificare l'impatto di questi effetti, già dimostrati molto importanti nelle dinamiche di rilassamento dell'elettrone in eccesso, sull'elettrone idratato. Ha anche rivelato una diffusione transitoria, un insolito, evento raro che non è presente nel regime classico. Mentre la diffusione non transitoria dell'elettrone solvatato è ottenuta mediante scambio di solvente seguito da un graduale spostamento della "nube di elettroni" o distribuzione della densità di spin, la diffusione transitoria è piuttosto un salto della densità di spin dalla cavità stabile a quella adiacente.
Mentre l'approccio della particella fantasma è stato applicato qui all'elettrone solvatato, potrebbe essere applicato anche a stati eccitati e quasiparticelle come polaroni, aprendo nuove opportunità per unire la teoria della struttura elettronica di alto livello con l'apprendimento automatico per ottenere simulazioni dinamiche altamente accurate a un prezzo moderato.