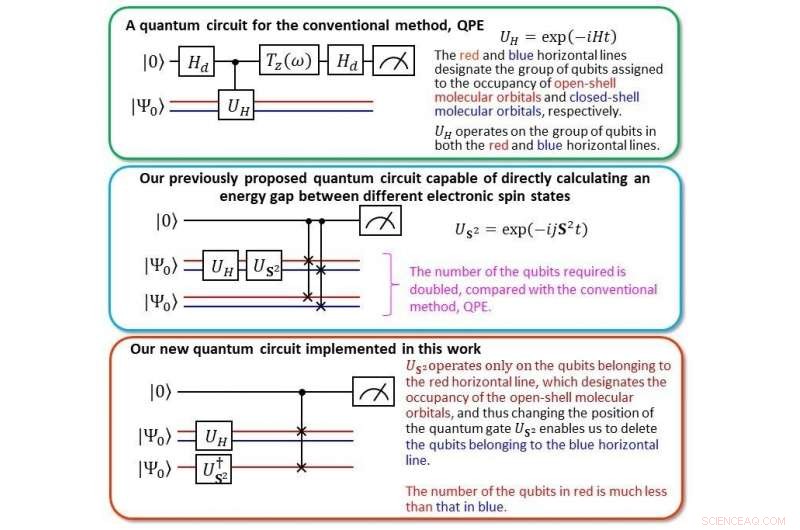
Confronto del nuovo circuito quantistico con il nostro precedente Credit:Kenji Sugisaki, Takeji Takui, Kazunobu Sato
I computer quantistici hanno ricevuto molta attenzione di recente poiché si prevede che risolvano determinati problemi che sono al di fuori delle capacità dei normali computer. Il principale di questi problemi è determinare gli stati elettronici di atomi e molecole in modo che possano essere utilizzati in modo più efficace in una varietà di settori, dai progetti di batterie agli ioni di litio alle tecnologie in silico nello sviluppo di farmaci. Un modo comune in cui gli scienziati hanno affrontato questo problema è calcolando le energie totali dei singoli stati di una molecola o atomo e quindi determinando la differenza di energia tra questi stati. In natura, molte molecole crescono in dimensione e complessità, e il costo per calcolare questo flusso costante è al di là della capacità di qualsiasi computer tradizionale o che attualmente stabilisce algoritmi quantistici. Perciò, previsioni teoriche delle energie totali sono state possibili solo se le molecole non sono dimensionabili e isolate dal loro ambiente naturale.
"Affinché i computer quantistici siano una realtà, i suoi algoritmi devono essere sufficientemente robusti da prevedere con precisione gli stati elettronici di atomi e molecole, come esistono in natura, " dichiarano Kenji Sugisaki e Takeji Takui della Graduate School of Science, Università della città di Osaka.
A dicembre 2020, Sugisaki e Takui, insieme ai loro colleghi, ha portato un team di ricercatori a sviluppare un algoritmo quantistico chiamato calcolatore dei parametri di accoppiamento di scambio bayesiano con funzioni d'onda a simmetria spezzata (BxB), che predice gli stati elettronici di atomi e molecole calcolando direttamente le differenze di energia. Hanno notato che le differenze di energia negli atomi e nelle molecole rimangono costanti, indipendentemente da quanto complessi e grandi diventino, nonostante le loro energie totali crescano man mano che le dimensioni del sistema. "Con BxB, abbiamo evitato la pratica comune di calcolare le energie totali e mirato direttamente alle differenze di energia, mantenere i costi di calcolo entro il tempo polinomiale, " affermano. "Da allora, il nostro obiettivo è stato quello di migliorare l'efficienza del nostro software BxB in modo che possa prevedere lo stato elettronico di atomi e molecole con precisione chimica."
Utilizzando i costi di calcolo di un noto algoritmo chiamato Quantum Phase Estimation (QPE) come benchmark, "abbiamo calcolato le energie di ionizzazione verticale di piccole molecole come CO, oh 2 , CN, F 2 , h 2 Oh, NH 3 entro 0,1 elettronvolt (eV) di precisione, " afferma la squadra, usando metà del numero di qubit, portando il costo di calcolo alla pari con QPE.
I loro risultati saranno pubblicati online nell'edizione di marzo del Journal of Physical Chemistry Letters .
L'energia di ionizzazione è una delle proprietà fisiche più fondamentali di atomi e molecole e un indicatore importante per comprendere la forza e le proprietà dei legami e delle reazioni chimiche. In breve, prevedere con precisione l'energia di ionizzazione ci consente di utilizzare prodotti chimici oltre la norma attuale. Nel passato, era necessario calcolare le energie degli stati neutri e ionizzati, ma con l'algoritmo quantistico BxB, l'energia di ionizzazione può essere ottenuta in un unico calcolo senza ispezionare le singole energie totali degli stati neutro e ionizzato. "Dalle simulazioni numeriche del circuito logico quantistico in BxB, abbiamo scoperto che il costo computazionale per leggere l'energia di ionizzazione è costante indipendentemente dal numero atomico o dalla dimensione della molecola, "afferma la squadra, "e che l'energia di ionizzazione può essere ottenuta con un'elevata precisione di 0,1 eV dopo aver modificato la lunghezza del circuito logico quantistico per essere inferiore a un decimo di QPE".
Con lo sviluppo dell'hardware dei computer quantistici, Sugisaki e Takui, insieme alla loro squadra, si aspettano che l'algoritmo quantistico BxB esegua calcoli energetici ad alta precisione per grandi molecole che non possono essere trattate in tempo reale con i computer convenzionali.