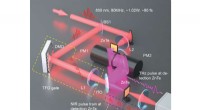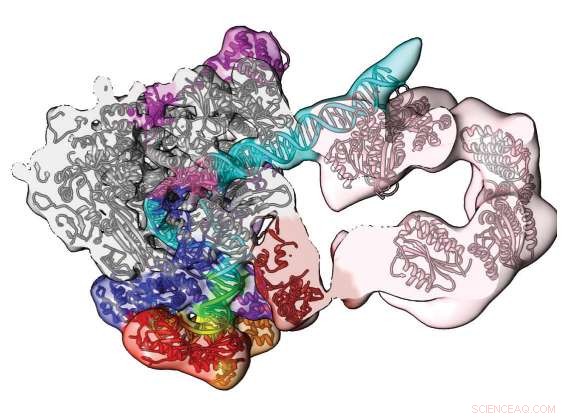
Il supercalcolo e la microscopia crioelettronica rivelano questa sezione del complesso di pre-iniziazione umano. La mappa e il modello della densità di conformazione aperta mostrano il percorso del DNA (blu/verde) e il suo impegno da parte del componente del fattore di trascrizione TFIIH (rosa). Ristampato con il permesso di Macmillan Publishers Ltd:He, Y. et al. Visualizzazione con risoluzione quasi atomica dell'apertura del promotore della trascrizione umana. Natura 533, 359-365 (2016).
Sembra uscito dai Borg in Star Trek. Robot di dimensioni nanometriche si autoassemblano per formare macchine biologiche che svolgono il lavoro che mantiene in vita. Eppure qualcosa del genere va avanti davvero.
Ogni cellula del nostro corpo - siano carne e sangue, cervello e tutto il resto - ha DNA identico, la scala contorta degli acidi nucleici codificata in modo univoco per ciascun organismo. Assemblaggi complessi che assomigliano a macchine molecolari prendono pezzi di DNA chiamati geni e creano una cellula cerebrale quando necessario, invece di, dire, una cellula ossea. Queste macchine molecolari sono così complesse, eppure così piccolo, che gli scienziati di oggi stanno appena iniziando a comprenderne la struttura e il funzionamento utilizzando gli ultimi microscopi e supercomputer. Le macchine molecolari biologiche potrebbero gettare le basi per lo sviluppo di cure per malattie come il cancro. Quanto piccolo si può vedere, e cosa si troverà?
La microscopia crioelettronica combinata con simulazioni al supercomputer hanno creato il miglior modello mai realizzato, con dettagli quasi a livello atomico, di una macchina molecolare vitale, il complesso di pre-iniziazione umana (PIC). Un team scientifico della Northwestern University, Laboratorio Nazionale di Berkeley, Università statale della Georgia, e UC Berkeley hanno pubblicato i loro risultati sul PIC maggio 2016 nella rivista Natura .
"Per la prima volta, sono state dettagliate le strutture dei complessi gruppi di molecole che aprono il DNA umano, ", ha affermato il coautore dello studio Ivaylo Ivanov, professore associato di chimica alla Georgia State University. Ivanov ha guidato il lavoro computazionale che ha modellato gli atomi delle diverse proteine che agiscono come ingranaggi della macchina molecolare PIC.
Il PIC trova geni associati alla produzione di una proteina specifica, come un anticorpo o un enzima. Lì il PIC separa i due filamenti di DNA e alimenta il filamento codificante all'enzima cavallo di battaglia RNA polimerasi II. Questo inizia la trascrizione, dove i bit di DNA vengono copiati dalla RNA polimerasi II in un singolo filamento di RNA messaggero. L'RNA si fa strada verso le "fabbriche proteiche" nella cellula chiamate ribosomi che li prendono come ordini per quale proteina produrre. Se il DNA è come il progetto di una nuova casa, Gli RNA sono istruzioni per gli "appaltatori" presso la stazione di lavoro dei ribosomi. Le proteine prodotte sono come le unghie, Di legno, Malta, e quasi tutto il resto della casa.
L'esperimento è iniziato con immagini di PIC accuratamente scattate. Sono stati realizzati da un gruppo guidato dalla coautrice dello studio Eva Nogales, professore nel Dipartimento di Biologia Molecolare e Cellulare dell'UC Berkeley e anche Senior Faculty Scientist presso il Lawrence Berkeley National Laboratory e Howard Hughes Medical Investigator.
Il gruppo di Nogales ha utilizzato la microscopia crioelettronica (crio-EM), una stella nascente nelle tecniche di laboratorio. Hanno congelato criogenicamente il PIC umano legato al DNA. Il congelamento lo ha mantenuto in un ambiente chimicamente attivo, ambiente quasi naturale. Poi l'hanno fulminato con fasci di elettroni. Grazie ai recenti progressi nella tecnologia dei rivelatori di elettroni diretti, cryo-EM può ora visualizzare a risoluzione quasi atomica strutture biologiche grandi e complicate che si sono dimostrate troppo difficili da cristallizzare. La tecnica di riferimento, Cristallografia a raggi X, richiede campioni cristallizzati, e cryo-EM evita questo difficile passaggio.
Oltre 1,4 milioni di "fermo fotogrammi" crio-EM di PIC sono stati elaborati utilizzando supercomputer presso il National Energy Research for Scientific Computing Center per setacciare il rumore di fondo e ricostruire mappe di densità tridimensionali che mostrano dettagli nella forma della molecola che non erano mai stati visto prima.
"Cryo-EM sta attraversando una grande espansione, così come tutti i software informatici utilizzati per generare sia le mappe di densità che per interpretarle come abbiamo fatto in questo studio, " Ha detto Nogales. "Ci consente di ottenere una risoluzione più elevata di più strutture in diversi stati in modo da poter descrivere non solo un'immagine di come appaiono, ma diverse immagini che mostrano come si stanno muovendo. Non vediamo un continuum, ma vediamo istantanee attraverso il processo di azione."
Gli scienziati dello studio hanno quindi costruito un modello accurato che ha dato un senso fisico alle mappe di densità di PIC utilizzando XSEDE, l'ambiente di scoperta della scienza e dell'ingegneria eXtream, finanziato dalla National Science Foundation. XSEDE consente agli scienziati di condividere in modo interattivo le risorse di elaborazione, dati e competenze attraverso un unico sistema virtuale. Il team di Ivaylo Ivanov ha eseguito oltre quattro milioni di ore centrali di simulazioni sul supercomputer Stampede presso il Texas Advanced Computing Center per modellare macchine molecolari complesse, compresi quelli per questo studio. Il più ampio lavoro di Ivanov sulla macchina molecolare include anche un'assegnazione XSEDE di 1,7 milioni di ore core sul supercomputer Comet presso il San Diego Supercomputing Center.
"Utilizzo le risorse XSEDE da più di 12 anni ormai, " Ivanov ha detto. "Senza la disponibilità di risorse XSEDE, tutta la nostra ricerca sarebbe stata molto più limitata in termini di sistemi che possiamo affrontare. Per noi, XSEDE è stato assolutamente essenziale."
L'obiettivo di tutto questo sforzo computazionale è produrre modelli atomici che raccontino la storia completa della struttura e della funzione del complesso proteico delle molecole. Per arrivarci, il team di Ivanov ha preso i dodici componenti dell'assieme PIC e ha creato modelli di omologia per ciascun componente che rappresentavano le loro sequenze di amminoacidi e la loro relazione con strutture 3D di proteine note simili.
Successivamente hanno approssimato le densità sperimentali che il team di Nogales ha trovato su una griglia. "Possiamo usare un metodo chiamato adattamento flessibile della dinamica molecolare, " ha spiegato Ivanov, "dove essenzialmente esegui una simulazione di dinamica molecolare. E usi la densità sperimentale per polarizzare gli atomi nella simulazione di dinamica molecolare per spostarti nelle regioni più dense della mappa EM. Questo è il processo di adattamento flessibile alla mappa EM".
Hanno perfezionato il modello con il pacchetto di raffinatezza cristallografica Phoenix. "Questa è una tecnica complementare che ci consente di posizionare le catene laterali e migliorare il modello in modo da poter catturare tutti i dettagli presenti nella mappa della densità, " disse Ivanov.
XSEDE era "assolutamente necessario" per questa modellazione, disse Ivanov. "Quando includiamo acqua e controioni oltre al complesso PIC in una scatola di simulazione di dinamica molecolare, otteniamo la dimensione del sistema di simulazione di oltre un milione di atomi. Non è possibile eseguirlo su una stazione di lavoro o anche su un cluster modesto. Per questo abbiamo davvero bisogno di un migliaio di core. In questo caso, siamo saliti a duemilaquarantotto core. E per questo avevamo bisogno dell'accesso a Stampede, " disse Ivanov.
Una delle intuizioni acquisite nello studio è un modello funzionante di come il PIC apre la doppia elica del DNA altrimenti stabile per la trascrizione. Nogales ha spiegato che si potrebbe immaginare una corda fatta di due fili attorcigliati l'uno intorno all'altro. Tieni un'estremità molto stretta. Afferra l'altro e ruotalo nella direzione opposta alla filettatura per sbrogliare il cavo. Fondamentalmente è così che fanno le macchine viventi che ci tengono in vita.
"Il DNA deve essere aperto e spostato nel sito attivo della polimerasi per codificare il primo nucleotide di RNA, " ha spiegato Nogales. "Il complesso di pre-iniziazione tiene i due filamenti del DNA molto strettamente insieme a un'estremità, in modo che non possano muoversi e non possano aprirsi. Dall'altra parte del PIC c'è una macchina che usa l'energia per spingere il DNA, attorcigliandolo nella direzione opposta in cui vengono infilati i due fili. E quando questo accade, tra i due lati, i fili si apriranno, ", ha detto Nogales.
Questo studio ha risolto la struttura di quella macchina molecolare che agisce come le dita che si attorcigliano, il fattore di trascrizione componente TFIIH. "TFIIH ha una sottounità di traslocazione, il cui ruolo è quello di spingere contemporaneamente il DNA verso il sito attivo della polimerasi e svolgere il DNA. Con la spinta e lo svolgimento combinati, effettivamente stai separando i due filamenti del DNA, " disse Ivanov.
Entrambi gli scienziati hanno affermato che stanno appena iniziando a comprendere a livello atomico la trascrizione, cruciale per l'espressione genica e, infine, per la malattia. "Molti stati patologici si verificano perché ci sono errori in quanto un certo gene viene letto e quanto una certa proteina con una certa attività è presente nella cellula, " ha detto Nogales. "Questi stati patologici potrebbero essere dovuti alla produzione eccessiva della proteina, o al contrario non abbastanza. È molto importante comprendere il processo molecolare che regola questa produzione in modo da poter comprendere lo stato della malattia".
"Questo lavoro illustra bene due principi generali che guideranno la scienza nei prossimi anni, " ha commentato Peter Preusch, funzionario del programma presso il National Institutes of Health (NIH). "Uno è l'applicazione di metodi ibridi:combinazioni di metodi biofisici tra cui cristallografia a raggi X e cryoEM insieme a metodi computazionali su larga scala per integrare informazioni su complessi molecolari più grandi. Due, c'è l'esigenza che la scienza di squadra attiri l'esperienza di più ricercatori per risolvere problemi che non possono essere affrontati da un singolo laboratorio che lavora da solo." Peter Preusch è il capo del ramo di biofisica, Divisione di Biologia Cellulare e Biofisica, Istituto Nazionale di Scienze Mediche Generali, NIH.
Sebbene questo lavoro fondamentale non produca direttamente cure, getta le basi per aiutarli a svilupparli in futuro, disse Ivanov. "Per capire la malattia, dobbiamo capire in primo luogo come funzionano questi complessi... Una collaborazione tra modellatori computazionali e biologi strutturali sperimentali potrebbe essere molto fruttuosa in futuro. "
Lo studio Nature Articles di maggio 2016 (DOI:10.1038/nature17970), "Visualizzazione a risoluzione quasi atomica dell'apertura del promotore della trascrizione umana, " è stato scritto da Yuan He, Lawrence Berkeley National Laboratory e ora alla Northwestern University; Chunli Yan e Ivaylo Ivanov, Università statale della Georgia; Jie Fang, Carla Inouye, Robert Tjian, Eva Nogales, UC Berkeley. Il finanziamento è venuto dal National Institute of General Medical Sciences (NIH) e dalla National Science Foundation.