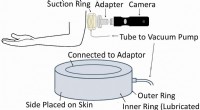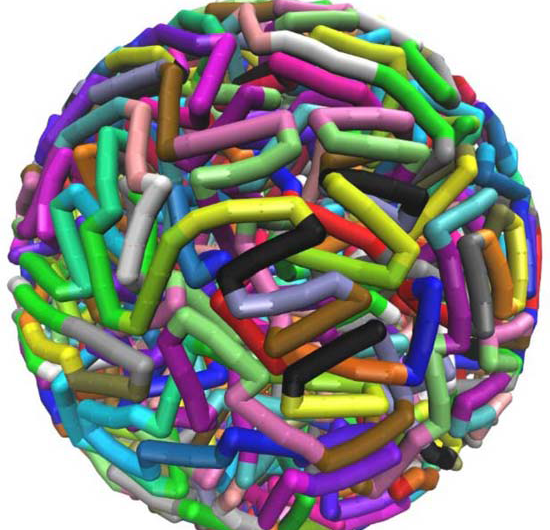
Specifico della sequenza, torsione indotta, configurazioni elastiche piegate, generato da simulazioni di dinamica molecolare su supercomputer presso il Texas Advanced Computing Center, aiutano a spiegare quanto lunghi filamenti di DNA possono stare in piccoli spazi. Credito:Christopher G. Myers, B. Montgomery Pettitt, Università del Texas Medical Branch
Un mistero biologico giace al centro di ciascuna delle nostre cellule, ovvero:come un metro di DNA può essere addensato nello spazio di un micron (o un milionesimo di metro) all'interno di ogni nucleo del nostro corpo.
I nuclei delle cellule umane non sono nemmeno il luogo biologico più affollato che conosciamo. Alcuni batteriofagi, virus che infettano e si replicano all'interno di un batterio, hanno un DNA ancora più concentrato.
"Come fa ad entrare lì dentro?" B. Montgomery (Monte) Pettitt, un biochimico e professore presso l'Università del Texas Medical Branch, chiede. "È un polimero carico. Come supera la repulsione alla sua densità cristallina liquida? Quanto ordine e disordine sono consentiti, e in che modo questo gioca un ruolo negli acidi nucleici?"
Utilizzando i supercomputer Stampede e Lonestar5 presso l'Università del Texas presso il Texas Advanced Computing Center (TACC) di Austin, Pettitt indaga su come il DNA dei fagi si ripiega in spazi iper-confinati.
Scrivendo nel numero di giugno 2017 del Journal of Computational Chemistry , ha spiegato come il DNA può superare sia la repulsione elettrostatica che la sua naturale rigidità.
La chiave per farlo? Perdite.
L'introduzione di forti torsioni o curve in configurazioni di DNA confezionato all'interno di un involucro sferico riduce significativamente le energie e le pressioni complessive della molecola, secondo Pettit.
Lui e i suoi collaboratori hanno usato un modello che deforma e attorciglia il DNA ogni 24 paia di basi, che è vicino alla lunghezza media prevista dalla sequenza del DNA del fago. L'introduzione di tali difetti persistenti non solo riduce l'energia di flessione totale del DNA confinato, ma riduce anche la componente elettrostatica dell'energia e della pressione.
"Mostriamo che un ampio insieme di configurazioni polimeriche è coerente con i dati strutturali, " lui e il collaboratore Christopher Myers, anche dell'Università del Texas Medical Branch, ha scritto.
Intuizioni come queste non possono essere acquisite rigorosamente in laboratorio. Richiedono supercomputer che fungono da microscopi molecolari, tracciare il movimento degli atomi e dei legami atomici su scale di lunghezza e tempo che non è possibile studiare con i soli esperimenti fisici.
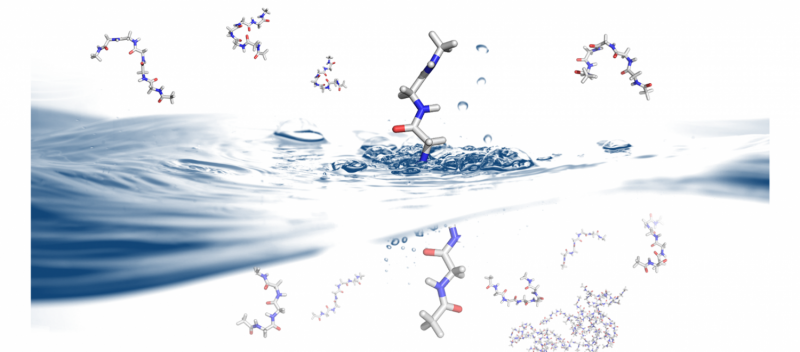
Come e perché le proteine si ripiegano è un problema che ha implicazioni per la progettazione e le terapie delle proteine. B. Montgomery Pettitt e il suo gruppo di ricerca presso l'Università del Texas Medical Branch utilizzano i supercomputer Stampede e Lonestar5 presso il Texas Advanced Computing Center per esplorare le dinamiche del ripiegamento delle proteine in soluzione. Credito:Christopher G. Myers, B. Montgomery Pettitt, Università del Texas Medical Branch
"Nel campo della biologia molecolare, c'è una meravigliosa interazione tra teoria, esperimento e simulazione, " ha detto Pettitt. " Prendiamo i parametri degli esperimenti e vediamo se sono d'accordo con le simulazioni e le teorie. Questo diventa il metodo scientifico per il modo in cui ora avanziamo le nostre ipotesi".
Problemi come quelli a cui è interessato Pettitt non possono essere risolti su un computer desktop o su un tipico cluster di campus, ma richiedono centinaia di processori di computer che lavorano in parallelo per imitare i minimi movimenti e le forze fisiche delle molecole in una cellula.
Pettitt è in grado di accedere ai supercomputer di TACC in parte grazie a un programma unico noto come Journal of Computational Chemistry iniziativa, che rende le risorse informatiche di TACC, competenze e formazione disponibili per i ricercatori all'interno delle 14 istituzioni dell'Università del Texas Systems.
"Ricerca computazionale, come quella del dottor Pettitt, che cerca di colmare la nostra comprensione del fisico, chimico, e infine fenomeni biologici, implica così tanti calcoli che è davvero accessibile solo su grandi supercomputer come i sistemi Stampede o Lonestar5 di TACC, " ha detto Brian Beck, un ricercatore di scienze della vita presso TACC.
"Avere a disposizione risorse di supercalcolo TACC è fondamentale per questo stile di ricerca, " ha detto Pettit.
TROVARE L'ORDINE IN PROTEINE DISORDINATE
Un altro fenomeno che ha a lungo interessato Pettitt è il comportamento delle proteine intrinsecamente disordinate (IDP) e dei domini intrinsecamente disordinati, dove parti di una proteina hanno una forma disordinata.
A differenza dei cristalli o del DNA altamente compresso nei virus, che hanno distinti, forme rigide, Gli sfollati interni "si piegano in un pasticcio appiccicoso, " secondo Pettitt. Eppure sono fondamentali per tutte le forme di vita.
Si ritiene che negli eucarioti (organismi le cui cellule hanno sottostrutture complesse come i nuclei), circa il 30% delle proteine ha un dominio intrinsecamente disordinato. Più del 60 percento delle proteine coinvolte nella segnalazione cellulare (processi molecolari che prendono segnali dall'esterno della cellula o attraverso le cellule che dicono alla cellula quali comportamenti attivare e disattivare in risposta) hanno domini disordinati. Allo stesso modo, L'80% delle proteine di segnalazione correlate al cancro ha regioni IDP, il che le rende molecole importanti da comprendere.
Tra gli IDP che Pettitt e il suo gruppo stanno studiando ci sono fattori di trascrizione nucleare. Queste molecole controllano l'espressione dei geni e hanno un dominio di segnalazione ricco di amminoacido flessibile, glicina.

Le immagini sopra mostrano le distribuzioni di densità media su 21 configurazioni di DNA ciascuna simulata per 100 nanosecondi di dinamica molecolare dopo la minimizzazione utilizzando a) configurazioni completamente elastiche e b) piegate, per confronto con c) Mappa di densità Cryo-EM da ricostruzioni fagiche asimmetriche di P22 con densità del capside rimossa graficamente. Credito:Christopher G. Myers, B. Montgomery Pettitt, Università del Texas Medical Branch
Il ripiegamento del dominio di segnalazione del fattore di trascrizione nucleare non è determinato dal legame idrogeno e dagli effetti idrofobici, come la maggior parte delle molecole proteiche, secondo Pettit. Piuttosto, quando le molecole più lunghe trovano troppe glicine in uno spazio, vanno oltre la loro solubilità e iniziano ad associarsi tra loro in modi insoliti.
"È come aggiungere troppo zucchero al tè, "Spiega Pettitt. "Non diventerà più dolce. Lo zucchero deve cadere dalla soluzione e trovare un partner, precipitando in un grumo".
Scrivendo in Scienza delle proteine nel 2015, ha descritto le simulazioni molecolari eseguite su Stampede che hanno aiutato a spiegare come e perché gli sfollati interni collassano in strutture simili a globuli.
Le simulazioni hanno calcolato le forze dalle interazioni carbonile (CO) dipolo-dipolo, ovvero le attrazioni tra l'estremità positiva di una molecola polare e l'estremità negativa di un'altra molecola polare. Ha determinato che queste interazioni sono più importanti nel collasso e nell'aggregazione di lunghi filamenti di glicina rispetto alla formazione di legami H.
"Dato che la spina dorsale è una caratteristica di tutte le proteine, Le interazioni CO possono anche svolgere un ruolo nelle proteine di sequenza non banale in cui la struttura è determinata dall'impaccamento interno e dagli effetti stabilizzanti dei legami H e delle interazioni CO-CO, " ha concluso.
La ricerca è stata resa possibile da un'allocazione del tempo di calcolo su Stampede attraverso l'Extreme Science and Engineering Discovery Environment (XSEDE), supportato dalla National Science Foundation.
Pettit, un campione di lunga data del supercalcolo, non usa solo le risorse TACC stesso. Incoraggia altri studiosi, compresi i suoi colleghi del Sealy Center for Structural Biology and Molecular Biophysics, usare anche i supercomputer.
"Il calcolo avanzato è importante per l'analisi dei dati e il raffinamento dei dati dagli esperimenti, Microscopia a raggi X ed elettronica, e informatica, " dice. "Tutti questi problemi hanno problemi di elaborazione dei dati di grandi dimensioni che possono essere risolti utilizzando l'elaborazione avanzata".
Quando si tratta di svelare i misteri della biologia sulle scale più piccole, niente batte un supercomputer gigante.