I ricercatori della Rice University e del Baylor College of Medicine hanno utilizzato simulazioni al computer per studiare il processo mediante il quale l'emoagglutinina aiuta i virus a invadere e infettare le cellule. I ricercatori ritengono che il dominio dello stelo della proteina si dispiega e si ripiega in una configurazione diversa quando viene attivato, ma si ferma per rilasciare un peptide di fusione nascosto che lega il virus alla cellula bersaglio. Clicca sull'immagine per una versione più grande. Credito:Xingcheng Lin
C'è un intoppo nell'oscillazione di una proteina che rilascia il virus dell'influenza. I ricercatori della Rice University e del Baylor College of Medicine ritengono che questo meccanismo possa essere un obiettivo utile per impedire al virus di infettare le cellule.
In un articolo negli Atti della National Academy of Sciences, il team Rice-Baylor guidato dal biofisico José Onuchic e dai biochimici Jianpeng Ma e Qinghua Wang approfondisce ulteriormente un complesso di glicoproteine che ha iniziato a definire in un articolo del 2014.
Quella proteina, emoagglutinina, si trova sulla superficie dei virus influenzali e li aiuta ad attaccarsi ea trasportarsi attraverso le membrane protettive delle cellule bersaglio.
Il documento inizia a definire il meccanismo che consente alla proteina di dispiegarsi e ripiegarsi in un attimo, cambiando la sua forma per esporre un peptide che attacca il virus a una cellula e inizia l'infezione. I ricercatori ritengono che i farmaci terapeutici possano utilizzare questo meccanismo per spegnere il virus.
"Questa proteina inizia in uno stato piegato e passa attraverso una trasformazione globale, ripiegando in uno stato completamente diverso, " disse Onuchic, co-direttore del Centro di Fisica Biologica Teorica (CTBP) di Rice. "Ma c'è una piccola parte al centro che l'evoluzione ha conservato."
Quel singolo residuo amminoacidico conservato è l'intoppo che fa fermare la proteina nel processo di ripiegamento. Consente a un peptide di fusione sepolto all'interno di legarsi alla cellula bersaglio e iniziare a infettarla. Senza la pausa, il ripiegamento sarebbe troppo veloce perché avvenga la rilegatura.
L'autore principale e ricercatore postdottorato di Rice Xingcheng Lin ha modellato quella parte della proteina, l'anello B del dominio HA2. HA2 si trova sotto un altro dominio, un berretto noto come HA1 che muta per sfuggire alle difese del passato. Lin ha spiegato che HA1 è un obiettivo comune per i farmaci antinfluenzali perché il dominio del cappuccio esposto è più accessibile del dominio protetto HA2.
Il problema è che HA1 muta costantemente per resistere ai farmaci, Egli ha detto. Ciò influenza l'efficacia dei vaccini antinfluenzali ogni anno. Lin e Onuchic hanno affermato che l'HA2 rappresenta un bersaglio migliore per i farmaci perché il meccanismo è altamente conservato dall'evoluzione.
"Se un farmaco prende di mira l'HA2, il dominio non può sfuggire effettuando mutazioni perché le mutazioni stesse lo renderebbero non funzionale, Lin ha detto. "Quel tipo di farmaco potrebbe diventare un vaccino universale".
HA2 è una struttura trimerica che, quando innescato da condizioni acide nell'ambiente vicino a una cellula bersaglio, si trasforma da un loop casuale a una bobina arrotolata. Anche con la pausa, si apre e si ripiega in una frazione di secondo, troppo veloce per essere visto dai microscopi. Ma una simulazione al computer del processo può essere rallentata.
Questa sembra essere una specialità del CTBP, che utilizza programmi che analizzano il panorama energetico delle proteine per prevedere come si piegheranno. Onuchic e i suoi colleghi sono pionieri nella teoria secondo cui le proteine ripiegate seguono un ordine ordinato, processo "incanalato" che dipende dall'energia intrinseca di ogni atomo della catena, ognuno dei quali cerca costantemente il suo stato energetico più basso. Se tutte le "perline" atomiche possono essere identificate, è possibile simulare il complesso processo di piegatura.
I ricercatori di Rice usano spesso modelli di proteine a grana grossa, un sottoinsieme di atomi che rappresentano il tutto, per prevedere come si piegheranno. Il nuovo studio era molto più ambizioso e si proponeva di prevedere il complesso svolgersi e ripiegarsi utilizzando non solo ogni atomo della catena ma anche ogni atomo nel suo ambiente liquido, disse Onuchic.
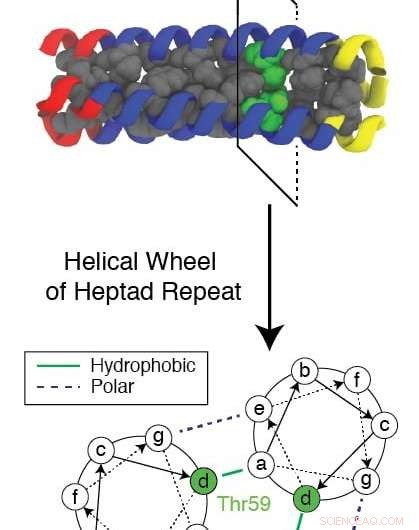
Un residuo conservato evolutivo noto come Thr59 interrompe il modello ripetuto formato da una proteina trimerica mentre si ripiega mentre aiuta un virus dell'influenza a infettare una cellula. I ricercatori della Rice University e del Baylor College of Medicine hanno utilizzato una complessa simulazione al computer per studiare il meccanismo e cercare nuovi bersagli per i farmaci per fermare l'influenza. Credito:Xingcheng Lin
Lin ha modellato 40 microsecondi (milionesimi di secondo) della transizione del dominio HA2 che rappresenta l'intero processo, che impiega 1,4 millisecondi (millesimi di secondo) per essere completata. Anche quel processo abbreviato ha richiesto due anni di tempo al computer per fornire risultati, Egli ha detto.
"Il dominio simulato è di circa 3, 000 atomi, ma quando l'ambiente compresa l'acqua, è contabilizzato, la simulazione totale comprende circa 100, 000 atomi, " Ha detto Onuchic. "È ancora un'enorme simulazione che richiedeva tecniche all'avanguardia".
Le teorie precedenti basate su immagini cristallografiche delle proteine prima e dopo hanno avanzato l'idea di un dominio caricato a molla che sembrava attaccarsi alla cellula bersaglio dopo la rimozione del cappuccio. Onuchic ha affermato che il modello completo di HA2 supporta un meccanismo diverso.
"Abbiamo scoperto che c'è un mucchio di energia che rende lo stato finale di HA2 molto più stabile dello stato iniziale, " ha detto. "Ma con il meccanismo a molla, la maggior parte dell'energia sarebbe già sprecata nel momento in cui forma la bobina arrotolata e lega la cellula e le membrane virali. Non lascerebbe alcuna energia per tirare insieme le membrane.
"Ecco perché abbiamo deciso di fare un calcolo completo del sistema:tutti gli atomi della proteina e tutta l'acqua, " Disse Onuchic. "È stato uno sforzo gigantesco".
Il residuo idrofilo conservato (che attira l'acqua), noto come Thr59, è di particolare interesse per i ricercatori non solo per il modo in cui interrompe il ripiegamento e consente al virus di attaccare, ma anche perché ha un gemello.
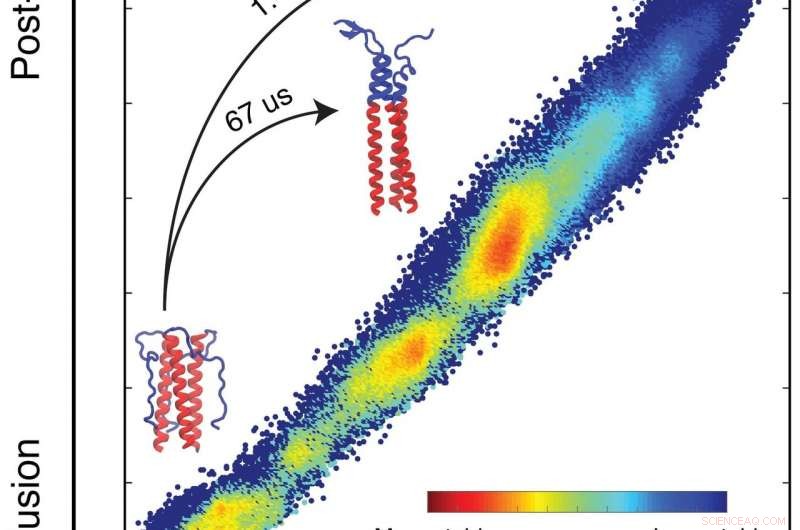
Una simulazione dei biofisici della Rice University ha dettagliato il profilo di energia libera che determina come una proteina che aiuta il virus dell'influenza a infettare le cellule svolge la sua missione. Le simulazioni prevedono come si piegherà una proteina in base alle energie intrinseche di ciascun atomo nel sistema. Le proteine formano anse e spirali mentre cercano il loro punto più basso, stati energetici più stabili (blu). Nel dominio studiato dai ricercatori, hanno trovato un intoppo che rallenta il processo di piegatura che consente il legame alla cellula bersaglio e presenta anche un'opportunità per nuovi vaccini di attaccare l'influenza. Clicca sull'immagine per una versione più grande. Credito:Xingcheng Lin
"Nell'intero albero evolutivo, questi virus si dividono in due gruppi, e la differenza sembra essere questo residuo, " Disse Onuchic. "Si sono divisi 1, 500 anni fa e in qualche modo, dopo questa separazione, sono completamente conservati. Non sono stati in grado di cambiare quel residuo, qualunque cosa accada, e crediamo che questo renda importante questo residuo".
L'attuale ricerca si è concentrata sul gruppo che incorpora Thr59 e causa il ceppo H3N2 responsabile dell'influenza di Hong Kong, disse Lin. L'altro residuo Met59, compare nel ceppo H1N1 che ha causato l'influenza spagnola.
"Abbiamo ancora molta strada da fare per capire l'intera proteina, " disse. "Ecco, abbiamo studiato solo un dominio di una proteina, e ce ne sono molti altri che sono molto importanti per la sua funzione."
"Ma quello che Xingcheng ha già fatto è un tour de force computazionale, Onuchic ha aggiunto. "Ha mostrato come questo particolare residuo rompe la simmetria elicoidale del dominio e lo rende abbastanza instabile da dare al peptide il tempo di afferrare le membrane".