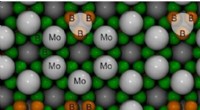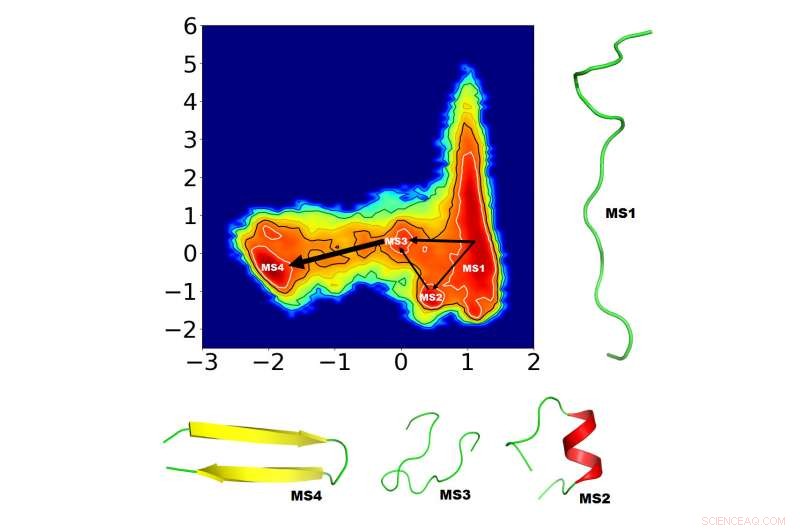
Gli scienziati cercano di comprendere meglio il ripiegamento delle proteine per curare le malattie da ripiegamento errato, ma questo processo incredibilmente complesso richiede algoritmi sofisticati per identificare i meccanismi di piegatura. I biofisici computazionali hanno proposto un nuovo modo per identificare i fattori più cruciali per il ripiegamento delle proteine. Hanno dimostrato il breve tempo di simulazione del loro approccio su una proteina piccola ma intrigante, "GB1 beta-tornante, " nel Giornale di Fisica Chimica . I quattro nuovi stati di piegatura intermedi (MS1-4) identificati dal team sono mostrati qui, insieme alle possibili vie di collegamento. Lo spessore delle frecce di interconnessione riflette la probabilità che si verifichi il percorso. Credito:Navjeet Ahalawat e Jagannath Mondal
I modelli di piegatura di una proteina li aiutano a svolgere i loro compiti dedicati. Come i veri "operatori" della cellula, anche una piccola alterazione nella struttura aminoacidica di una proteina può causare un mal ripiegamento e ostacolare la funzionalità della proteina o causare malattie. Ad esempio, se tau, una proteina che aiuta a stabilizzare la struttura delle cellule cerebrali, è piegato male, può formare grovigli tau, che sono comunemente osservati nei pazienti di Alzheimer.
Gli scienziati cercano di comprendere meglio il ripiegamento delle proteine per curare le malattie da ripiegamento errato, ma questo processo incredibilmente complesso richiede algoritmi sofisticati per identificare i meccanismi di piegatura. I biofisici computazionali del Tata Institute of Fundamental Research di Hyderabad (TIFR-H) hanno proposto un nuovo modo per identificare i fattori più cruciali per il ripiegamento delle proteine. Hanno dimostrato il breve tempo di simulazione del loro approccio su una proteina piccola ma intrigante, "GB1 beta-tornante, " nel Giornale di Fisica Chimica , da AIP Publishing.
"Combinando un metodo noto come "analisi dei componenti indipendenti basata sulla struttura del tempo" (TICA) con brevi simulazioni di dinamica molecolare, abbiamo trovato quattro stati di piegatura intermedi fisicamente significativi, non osservato in precedenza, e ha mostrato stati elicoidali che di solito non possono essere rilevati con altri metodi, " disse Navjeet Ahalawat, un autore sulla carta.
Ogni atomo di una proteina può ripiegarsi in tre dimensioni, ma con milioni di atomi presenti anche in proteine semplici, il compito di comprendere la combinazione di piegatura collettiva diventa contorto. Gli scienziati hanno considerato i diversi fattori che influenzano il ripiegamento delle proteine, come il legame idrogeno, e li ha combinati in descrizioni generali chiamate variabili collettive (CV). Però, con molti potenziali fattori, gli scienziati non hanno un buon modo per trovare CV che descrivano in modo appropriato un processo fattibile.
"Ci sono molti modi in cui le proteine possono passare da uno stato dispiegato a uno stato piegato, quindi la cosa più difficile è decidere da dove cominciare, " disse Ahalawat. Jagannath Mondal, un altro autore sulla carta, ha aggiunto che è stato facile "perdersi nei dati".
Il team ha deciso di studiare la forcina sporgente esternamente della proteina GB1 a causa dell'ampio corpo di lavoro esistente e di molte potenziali possibilità di ripiegamento già stimate nei CV passati. Ahalawat e Mondal hanno preso una serie di CV GB1 esistenti come CV costitutivi e li hanno combinati linearmente utilizzando TICA per identificare una coppia di CV "ottimizzati". Quindi, hanno inserito i CV ottimizzati nel modello di stato di Markov e hanno identificato quattro stati di piegatura intermedi insieme ai possibili percorsi di connessione.
"Noi abbiamo chiesto, quali sono le caratteristiche stimate in precedenza per questa particolare proteina che potrebbe davvero svolgere un ruolo chiave nel sistema? E possiamo trovare la giusta combinazione di condizioni?" Ha detto Ahalawat. "Nel nostro lavoro ora possiamo stabilire quantitativamente se quella caratteristica è rilevante per il processo".
"Utilizzando brevi simulazioni, abbiamo trovato il peso che è veramente necessario utilizzare in una combinazione, e questo dà il giusto schema di piegatura per una proteina, " Mondal ha aggiunto. "È un modo davvero economico per capire il ripiegamento delle proteine".
Nel loro metodo, i dati di studi precedenti sono necessari per identificare i CV ottimali. Il team prevede che la loro tecnica possa essere utilizzata per scoprire il meccanismo interno del ripiegamento delle proteine sane per correggere le malattie che causano il mal ripiegamento delle proteine. Vogliono anche sviluppare ulteriormente il loro metodo di ottimizzazione del CV e applicarli nel riconoscimento biomolecolare e nella scoperta di farmaci. "In futuro abbiamo in programma di incorporare metodi non lineari, utilizzando tecniche di deep learning basate su reti neurali per migliorare il nostro modello, "ha detto Ahalawat.